 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Shuen-Ei Chen | + 3446 word(s) | 3446 | 2021-09-26 05:05:09 | | | |
| 2 | Bruce Ren | -13 word(s) | 3433 | 2021-09-28 03:21:03 | | |
Video Upload Options
Heme oxygenases (HOs) act on heme degradation to produce carbon monoxide (CO), free iron, ferritin, and biliverdin. Upregulation of cellular HO-1 levels is signature of oxidative stress for its downstream effects particularly under pro-oxidative status. Subcellular traffics of HO-1 to different organelles constitute a network of interactions compromising a variety of effectors such as pro-oxidants, ROS, mitochondrial enzymes, and nucleic transcription factors. Some of the compartmentalized HO-1 have been demonstrated as functioning in the progression of cancer. Emerging data show the multiple roles of HO-1 in tumorigenesis from pathogenesis to the progression to malignancy, metastasis, and even resistance to therapy. However, the role of HO-1 in tumorigenesis has not been systematically addressed.
1. Heme Oxygenases (HOs) and Oxidative Stress
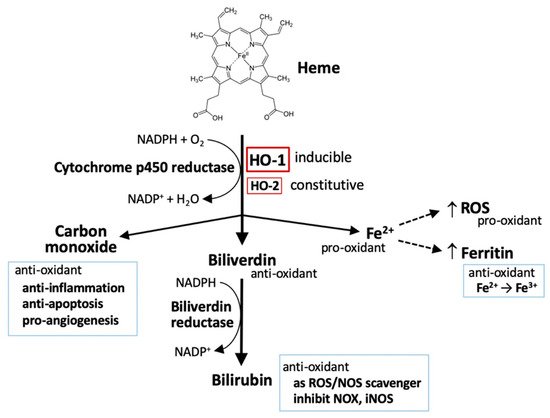
2. The Metabolism of Heme
2.1. Heme
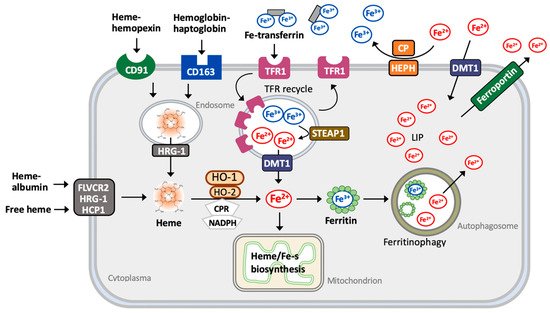
2.2. Iron
2.3. Carbon Monoxide (CO)
2.4. Bilirubin and Biliverdin
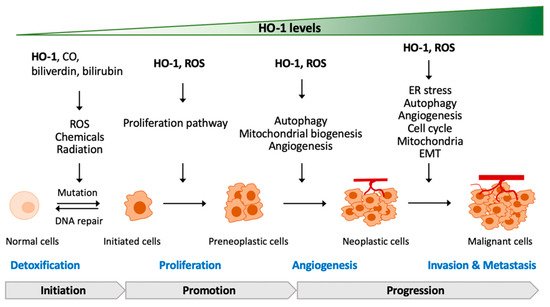
3. The Contradictory Role of HO-1 in Tumorigenesis
3.1. HO-1 Deficiency or Mutation in Tumorigenesis
3.2. HO-1-Regulated Proliferation and Development of Cancer Cells
3.3. HO-1-Regulated Angiogenesis of Cancer Cells
3.4. HO-1-Regulated Metastasis of Cancer Cells
References
- Tenhunen, R.; Marver, H.; Pimstone, N.R.; Trager, W.F.; Cooper, D.Y.; Schmid, R. Enzymatic degradation of heme. Oxygenative cleavage requiring cytochrome P-450. Biochemistry 1972, 11, 1716–1720.
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650.
- Kutty, R.K.; Kutty, G.; Rodriguez, I.R.; Chader, G.J.; Wiggert, B. Chromosomal localization of the human heme oxygenase genes: Heme oxygenase-1 (HMOX1) maps to chromosome 22q12 and heme oxygenase-2 (HMOX2) maps to chromosome 16p13.3. Genomics 1994, 20, 513–516.
- Munoz-Sanchez, J.; Chanez-Cardenas, M.E. A review on hemeoxygenase-2: Focus on cellular protection and oxygen response. Oxid. Med. Cell Longev. 2014, 2014, 604981.
- Hayashi, S.; Omata, Y.; Sakamoto, H.; Higashimoto, Y.; Hara, T.; Sagara, Y.; Noguchi, M. Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene 2004, 336, 241–250.
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Cell Biol. 2020, 21, 363–383.
- Schieber, M.; Chandel, N. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462.
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell Mol. Life Sci. 2016, 73, 3221–3247.
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Invest. 1999, 103, 129–135.
- Radhakrishnan, N.; Yadav, S.P.; Sachdeva, A.; Pruthi, P.K.; Sawhney, S.; Piplani, T.; Wada, T.; Yachie, A. Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J. Pediatr. Hematol. Oncol. 2011, 33, 74–78.
- Yachie, A. Heme oxygenase-1 deficiency and oxidative stress: A review of 9 independent human cases and animal models. Int. J. Mol. Sci. 2021, 22, 1514.
- Poss, K.D.; Tonegawa, S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. USA 1997, 94, 10919–10924.
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072.
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-accelerates Erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403.
- Chang, L.C.; Chiang, S.K.; Chen, S.E.; Yu, Y.L.; Chou, R.H.; Chang, W.C. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018, 416, 124–137.
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayır, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355.
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 induction in cancer progression: A matter of cell adaptation. Antioxidants 2017, 6, 29.
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A dual role of heme oxygenase-1 in cancer cells. Int. J. Mol. Sci. 2018, 20, 39.
- Gozzelino, R.; Soares, M.P. Coupling heme and iron metabolism via ferritin H chain. Antioxid. Redox Signal. 2014, 20, 1754–1768.
- Larsen, R.; Gouveia, Z.; Sorares, M.P.; Gozzelino, R. Heme cytotoxicity and the pathogenesis of immune-mediated inflammatory diseases. Front Pharmacol. 2012, 3, 77.
- Camaschella, C.; Nai, A.; Silvestri, L. Iron metabolism and iron disorders revisited in hepcidin era. Haematologica 2020, 105, 260–272.
- Broxmeyer, H.E.; Cooper, S.; Levi, S.; Arosio, P. Mutated recombinant human heavy-chain ferritins and myelosuppression in vitro and in vivo: A link between ferritin ferroxidase activity and biological function. Proc. Natl. Acad. Sci. USA 1991, 88, 770–774.
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytoxic effects of circulating heme. Blood 2002, 100, 879–887.
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242.
- Kharitonov, V.G.; Sharma, V.S.; Pilz, R.B.; Magde, D.; Koesling, D. Basis of guanylate cyclase activation by carbon monoxide. Proc. Natl. Acad. Sci. USA 1995, 92, 2568–2571.
- Motterlini, R.; Otterbein, L.E. The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 2010, 9, 728–743.
- Kim, K.M.; Pae, H.O.; Zheng, M.; Park, R.; Kim, Y.M.; Chung, H.T. Carbon monoxide induces heme oxygenase-1 via activation of protein kinase R-like endoplasmic reticulum kinase and inhibits endothelial cell apoptosis triggered by endoplasmic reticulum stress. Circ. Res. 2007, 101, 919–927.
- Brouard, S.; Otterbein, L.E.; Anrather, J.; Tobiasch, E.; Bach, F.H.; Choi, A.M.; Soares, M.P. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J. Exp. Med. 2000, 192, 1015–1026.
- Zhang, X.; Shan, P.; Alam, J.; Fu, X.Y.; Lee, P.J. Carbon monoxide differentially modulates STAT1 and STAT3 and inhibits apoptosis via a phosphatidylinositol 3-kinase/Akt and p38 kinase-dependent STAT3 pathway during anoxia-reoxygenation injury. J. Biol. Chem. 2005, 280, 8714–8721.
- Al-Owais, M.M.; Scragg, J.L.; Dallas, M.L.; Boycott, H.E.; Warburton, P.; Chakrabarty, A.; Boyle, J.P.; Peers, C. Carbon monoxide mediates the anti-apoptotic effects of heme oxygenase-1 in medulloblastoma DAOY cells via K+ channel inhibition. J. Biol. Chem. 2012, 287, 24754–24764.
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Tao Lu, H.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M. Carbon monoxide has anti-inflammatory effects involving the mitogen- activated protein kinase pathway. Nat. Med. 2000, 6, 422–428.
- Boczkowski, J.; Poderoso, J.J.; Motterlini, R. CO-metal interaction: Vital signaling from a lethal gas. Trends Biochem. Sci. 2006, 31, 614–621.
- Ryter, S.W.; Choi, A.M. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl. Res. 2016, 167, 7–34.
- Ryter, S.W.; Ma, K.C.; Choi, A.M.K. Carbon monoxide in lung cell physiology and disease, Am. J. Physiol. Cell Physiol. 2018, 314, C211–C227.
- Almeida, A.S.; Figueiredo-Pereira, C.; Vieira, H.L.A. Carbon monoxide and mitochondria—Modulation of cell metabolism, redox response and cell death. Front. Physiol. 2015, 6, 33.
- Sedlak, T.W.; Snyder, S.H. Bilirubin benefits: Cellular protection by a biliverdin reductase antioxidant cycle. Pediatrics 2004, 113, 1776–1782.
- Kwak, J.Y.; Takeshige, K.; Cheung, B.S.; Minakami, S. Bilirubin inhibits the activation of superoxide-producing NADPH oxidase in a neutrophil cell-free system. Biochim. Biophys. Acta 1991, 1076, 369–373.
- Lanone, S.; Bloc, S.; Foresti, R.; Almolki, A.; Taille, C.; Callebert, J.; Conti, M.; Goven, D.; Aubier, M.; Dureuil, B.; et al. Bilirubin decreases nos2 expression via inhibition of NAD(P)H oxidase: Implications for protection against endotoxic shock in rats. FASEB J. 2005, 19, 1890–1892.
- Jansen, T.; Daiber, A. Direct antioxidant properties of bilirubin and biliverdin. Is there a role for biliverdin reductase? Front. Pharmacol. 2012, 3, 30.
- Weinberg, F.; Ramnath, N.; Nagrath, D. Reactive oxygen species in the tumor microenvironment: An overview. Cancers 2019, 11, 1191.
- Mascaro, M.; Alonso, E.N.; Alonso, E.G.; Lacunza, E.; Curino, A.C.; Facchinetti, M.M. Nuclear localization of heme oxygenase-1 in pathophysiological conditions: Does it explain the dual role in cancer? Antioxidants 2021, 10, 87.
- Jozkowicz, A.; Was, H.; Dulak, J. Heme oxygenase-1 in tumors: Is it a false friend? Antioxid. Redox. Signal. 2007, 9, 2099–2117.
- Luu Hoang, K.N.; Anstee, J.E.; Arnold, J.N. The diverse roles of heme oxygenase-1 in tumor progression. Front. Immunol. 2021, 12, 658315.
- Otterbein, L.E.; Hedblom, A.; Harris, C.; Csizmadia, E.; Gallo, D.; Wegiel, B. Heme oxygenase-1 and carbon monoxide modulate DNA repair through ataxia- telangiectasia mutated (ATM) protein. Proc. Natl. Acad. Sci USA 2011, 108, 14491–14496.
- Barikbin, R.; Berkhout, L.; Bolik, J.; Schmidt-Arras, D.; Ernst, T.; Ittrich, H.; Adam, G.; Parplys, A.; Casar, C.; Krech, T.; et al. Early heme oxygenase 1 induction delays tumour initiation and enhances DNA damage repair in liver macrophages of Mdr2(−/−) mice. Sci. Rep. 2018, 8, 16238.
- Seiwert, N.; Wecklein, S.; Demuth, P.; Hasselwander, S.; Kemper, T.A.; Schwerdtle, T.; Brunner, T.; Fahrer, J. Heme oxygenase 1 protects human colonocytes against ROS formation, oxidative DNA damage and cytotoxicity induced by heme iron, but not inorganic iron. Cell Death Dis. 2020, 11, 787.
- Zhu, W.; Xu, J.; Ge, Y.; Cao, H.; Ge, X.; Luo, J.; Xue, J.; Yang, H.; Zhang, S.; Cao, J. Epigallocatechin-3-gallate (EGCG) protects skin cells from ionizing radiation via heme oxygenase-1 (HO-1) overexpression. J. Radiat. Res. 2014, 55, 1056–1065.
- Jin, J.; Wang, D.; Xiao, H.; Wei, H.; Matunda, C.; Zhang, H.; Li, X.; Wang, C.; Zou, C.; Gao, X.; et al. Enhancement of DEN-induced liver tumorigenesis in heme oxygenase-1 G143H mutant transgenic mice. Biochem. Biophys. Res. Commun. 2016, 481, 169–175.
- He, J.Z.; Ho, J.J.D.; Gingerich, S.; Courtman, D.W.; Marsden, P.A.; Ward, M.E. Enhanced translation of heme oxygenase-2 preserves human endothelial cell viability during hypoxia. J. Biol. Chem. 2010, 285, 9452–9461.
- Gandini, N.A.; Fermento, M.E.; Salomon, D.G.; Blasco, J.; Patel, V.; Gutkind, J.S.; Molinolo, A.A.; Facchinetti, M.M.; Curino, A.C. Nuclear localization of heme oxygenase-1 is associated with tumor progression of head and neck squamous cell carcinomas. Exp. Mol. Pathol. 2012, 93, 237–245.
- Sacca, P.; Meiss, R.; Casas, G.; Mazza, O.; Calvo, J.C.; Navone, N.; Vazquez, E. Nuclear translocation of haeme oxygenase-1 is associated to prostate cancer. Br. J. Cancer. 2007, 97, 1683–1689.
- Talieri, M.; Papadopoulou, S.; Scorilas, A.; Xynopoulos, D.; Arnogianaki, N.; Plataniotis, G.; Yotis, J.; Agnanti, N. Cathepsin B and cathepsin D expression in the progression of colorectal adenoma to carcinoma. Cancer Lett. 2004, 205, 97–106.
- Chan, A.T.; Baba, Y.; Shima, K.; Nosho, K.; Chung, D.C.; Hung, K.E.; Mahmood, U.; Madden, K.; Poss, K.; Ranieri, A.; et al. Cathepsin B expression and survival in colon cancer: Implications for molecular detection of neoplasia. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2777–2785.
- Gandini, N.A.; Fermento, M.E.; Salomon, D.G.; Obiol, D.J.; Andres, N.C.; Zenklusen, J.C.; Arevalo, J.; Blasco, J.; Lopez Romero, A.; Facchinetti, M.M.; et al. Heme oxygenase-1 expression in human gliomas and its correlation with poor prognosis in patients with astrocytoma. Tumour Biol. 2014, 35, 2803–2815.
- Mayerhofer, M.; Florian, S.; Krauth, M.T.; Aichberger, K.J.; Bilban, M.; Marculescu, R.; Printz, D.; Fritsch, G.; Wagner, O.; Selzer, E.; et al. Identification of heme oxygenase-1 as a novel BCR/ABL-dependent survival factor in chronic myeloid leukemia. Cancer Res. 2004, 64, 3148–3154.
- Balan, M.; Chakraborty, S.; Flynn, E.; Zurakowski, D.; Pal, S. Honokiol inhibits c-Met-HO-1 tumor-promoting pathway and its cross-talk with calcineurin inhibitor-mediated renal cancer growth. Sci. Rep. 2017, 7, 5900.
- Lien, G.S.; Wu, M.S.; Bien, M.Y.; Chen, C.H.; Lin, C.H.; Chen, B.C. Epidermal growth factor stimulates nuclear factor-kappaB activation and heme oxygenase-1 expression via c-Src, NADPH oxidase, PI3K, and Akt in human colon cancer cells. PLoS ONE 2014, 9, e104891.
- Alaluf, E.; Vokaer, B.; Detavernier, A.; Azouz, A.; Splittgerber, M.; Carrette, A.; Boon, L.; Libert, F.; Soares, M.; Le Moine, A.; et al. Heme oxygenase 1 orchestrates the immunosuppressive program of tumor-associated macrophages. JCI Insight 2020, 5, e133929.
- Sacco, A.; Battaglia, A.M.; Botta, C.; Aversa, I.; Mancuso, S.; Costanzo, F.; Biamonte, F. Iron metabolism in the tumor microenvironment-implications for anti-cancer immune response. Cells 2021, 10, 303.
- Nemeth, Z.; Li, M.; Csizmadia, E.; Dome, B.; Johansson, M.; Persson, J.L.; Seth, P.; Otterbein, L.; Wegiel, B. Heme oxygenase-1 in macrophages controls prostate cancer progression. Oncotarget 2015, 6, 33675–33688.
- Lugano, R.; Ramachandran, M.; Dimberg, A. tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell Mol. Life Sci. 2020, 7, 1745–1770.
- Sunamura, M.; Duda, D.G.; Ghattas, M.H.; Lozonschi, L.; Motoi, F.; Yamauchi, J.; Matsuno, S.; Shibahara, S.; Abraham, N.G. Heme oxygenase-1 accelerates tumor angiogenesis of human pancreatic cancer. Angiogenesis 2003, 6, 15–24.
- Deshane, J.; Chen, S.; Caballero, S.; Grochot-Przeczek, A.; Was, H.; Li Calzi, S.; Lach, R.; Hock, T.D.; Chen, B.; Hill-Kapturczak, N.; et al. Stromal cell-derived factor 1 promotes angiogenesis via a heme oxygenase 1-dependent mechanism. J. Exp. Med. 2007, 204, 605–618.
- Loboda, A.; Jazwa, A.; Grochot-Przeczek, A.; Rutkowski, A.J.; Cisowski, J.; Agarwal, A.; Jozkowicz, A.; Dulak, J. Heme oxygenase-1 and the vascular bed: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal 2008, 10, 1767–1812.
- Birrane, G.; Li, H.; Yang, S.; Tahado, S.D.; Seng, S. Cigarette smoke induces nuclear translocation of heme oxygenase 1 (HO-1) in prostate cancer cells: Nuclear HO-1 promotes vascular endothelial growth factor secretion. Int. J. Oncol. 2013, 42, 1919–1928.
- Cheng, C.C.; Guan, S.S.; Yang, H.J.; Chang, C.C.; Luo, T.Y.; Chang, J.; Ho, A.S. Blocking heme oxygenase-1 by zine protoporphyrin reduces tumor hypoxia-mediated VEGF release and inhibits tumor angiogenesis as a potential therapeutic agent against colorectal cancer. J. Biomed. Sci. 2016, 23, 18.
- Bauer, A.; Mylroie, H.; Thornton, C.C.; Calay, D.; Birdsey, G.M.; Kiprianos, A.P.; Wilson, G.K.; Soares, M.P.; Yin, X.; Mayr, M.; et al. Identification of cyclins A1, E1 and vimentin as downstream targets of heme oxygenase-1 in vascular endothelial growth factor-mediated angiogenesis. Sci. Rep. 2016, 6, 29417.
- Maamoun, H.; Zachariah, M.; McVey, J.H.; Green, F.R.; Agouni, A. Heme oxygenase (HO)-1 induction prevents endoplasmic reticulum stress-mediated endothelial cell death and impaired angiogenic capacity. Biochem. Pharmacol. 2017, 127, 46–59.
- Choi, Y.K.; Kim, C.K.; Lee, H.; Jeoung, D.; Ha, K.S.; Kwon, Y.G.; Kim, K.W.; Kim, Y.M. Carbon monoxide promotes VEGF expression by increasing HIF-1alpha protein level via two distinct mechanisms, translational activation and stabilization of HIF-1alpha protein. J. Biol. Chem. 2010, 285, 32116–32125.
- Lin, H.H.; Chiang, M.T.; Chang, P.C.; Chau, L.Y. Myeloid heme oxygenase-1 promotes metastatic tumor colonization in mice. Cancer Sci. 2015, 106, 299–306.
- Was, H.; Cichon, T.; Smolarczyk, R.; Rudnicka, D.; Stopa, M.; Chevalier, C.; Leger, J.J.; Lackowska, B.; Grochot, A.; Bojkowska, K.; et al. Overexpression of heme oxygenase-1 in murine melanoma: Increased proliferation and viability of tumor cells, decreased survival of mice. Am. J. Pathol. 2006, 169, 2181–2198.
- Skrzypek, K.; Tertil, M.; Golda, S.; Ciesla, M.; Weglarczyk, K.; Collet, G.; Guichard, A.; Kozakowska, M.; Boczkowski, J.; Was, H.; et al. Interplay between heme oxygenase-1 and miR-378 affects non-small cell lung carcinoma growth, vascularization, and metastasis. Antioxid. Redox Signal. 2013, 19, 644–660.
- Brabletz, T.; Kalluri, R.; Angela, N.; Weinberg, R.A. EMT in cancer. Nature Rev. Cancer 2018, 18, 128–134.
- Castruccio Castracani, C.; Longhitano, L.; Distefano, A.; Di Rosa, M.; Pittala, V.; Lupo, G.; Caruso, M.; Corona, D.; Tibullo, D.; Li Volti, G. Heme oxygenase-1 and carbon monoxide regulate growth and progression in glioblastoma cells. Mol. Neurobiol. 2020, 57, 2436–2446.
- Zhao, Z.; Zhao, J.; Xue, J.; Zhao, X.; Liu, P. Autophagy inhibition promotes epithelial-mesenchymal transition through ROS/HO-1 pathway in ovarian cancer cells. Am. J. Cancer Res. 2016, 6, 2162–2177.
- Chang, Y.J.; Chen, W.Y.; Huang, C.Y.; Liu, H.H.; Wei, P.L. Glucose-regulated protein 78 (GRP78) regulates colon cancer metastasis through EMT biomarkers and the NRF-2/HO-1 pathway. Tumour Biol. 2015, 36, 1859–1869.
- Wang, X.; Ye, T.; Xue, B.; Yang, M.; Li, R.; Xu, X.; Zeng, X.; Tian, N.; Bao, L.; Huang, Y. Mitochondrial GRIM-19 deficiency facilitates gastric cancer metastasis through oncogenic ROS-NRF2-HO-1 axis via a NRF2-HO-1 loop. Gastric Cancer 2021, 24, 117–132.
- Yang, P.S.; Hsu, Y.C.; Lee, J.J.; Chen, M.J.; Huang, S.Y.; Cheng, S.P. Heme Oxygenase-1 inhibitors induce cell cycle arrest and suppress tumor growth in thyroid cancer cells. Int. J. Mol. Sci. 2018, 19, 2502.
- Dey, S.; Sayers, C.M.; Verginadis, I.I.; Lehman, S.L.; Cheng, Y.; Cerniglia, G.J.; Tuttle, S.W.; Feldman, M.D.; Zhang, P.J.; Fuchs, S.Y.; et al. ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J. Clin. Investig. 2015, 125, 2592–2608.
- Tertil, M.; Golda, S.; Skrzypek, K.; Florczyk, U.; Weglarczyk, K.; Kotlinowski, J.; Maleszewska, M.; Czauderna, S.; Pichon, C.; Kieda, C.; et al. Nrf2-heme oxygenase-1 axis in mucoepidermoid carcinoma of the lung: Antitumoral effects associated with down-regulation of matrix metalloproteinases. Free Radic. Biol. Med. 2015, 89, 147–157.
- Li, Q.; Liu, Q.; Cheng, W.; Wei, H.; Jiang, W.E.F.; Yu, Y.; Jin, J.; Zou, C. Heme oxygenase-1 inhibits tumor metastasis mediated by notch1 pathway in murine mammary carcinoma. Oncol. Res. 2019, 27, 643–651.


