1. Heme Oxygenases (HOs) and Oxidative Stress
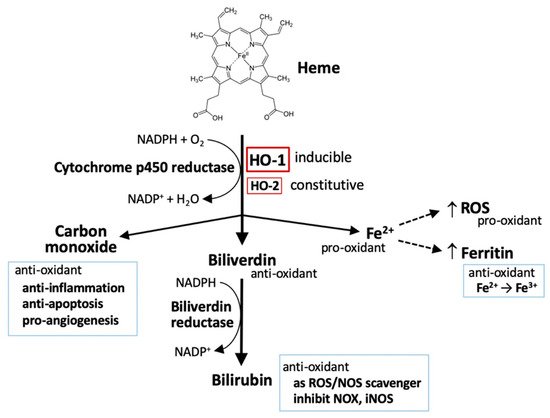
HOs are the key-limiting enzymes in heme degradation leading to carbon monoxide (CO), ferrous iron, and biliverdin products. Biliverdin is then rapidly converted to bilirubin by biliverdin reductase (
Figure 1)
[1][2][1,2]. HOs are expressed in a variety of cell types, rendering their broad contribution to cell functions. Currently, three mammalian HO isoforms are identified, namely HO-1, HO-2, and HO-3. HO-1 and HO-2 are identified in human and rats. HO-3 is only found in rats. HO-1 is inducible, synthetically enhanced by pro-oxidant stimuli. HO-1 is encoded by
HMOX1 with a molecular weight of around 32 kDa (also called heat shock protein 32; HSP32)
[3].
HMOX2 is constitutively expressed to encode a 36 kDa HO-2 protein, mainly functioning to maintain the basal heme metabolism and may also play a role in inflammatory responses
[4].
HMOX3 is distinguished as a pseudogene and has no transcript form
[5].
Figure 1. HO-1 and heme metabolism. Heme is degraded by heme oxygenases (HOs), generating biliverdin, carbon monoxide, and ferrous iron (Fe2+). Biliverdin is subsequently converted to bilirubin by biliverdin reductase. Both biliverdin and bilirubin act as anti-oxidants by scavenging or neutralizing reactive oxygen species (ROS). Carbon monoxide, a gaseous product, functions in signaling transduction, including the vasodilation of blood vessels, production of anti-inflammatory cytokines, upregulation of anti-apoptotic effectors, and promotion of angiogenesis. Ferrous iron (Fe2+) possesses pro-oxidant activity. However, activation of heme oxygenase-1 (HO-1) can upregulate ferritin expression, which binds to ferrous iron and detoxifies its pro-oxidant effect. Ferrous iron increases ROS generation via the Fenton reaction.
Oxidative stress is referred as a pro-oxidative circumstance, occurring at the imbalance between oxidants and antioxidants in favor of the oxidants, which has been implicated in governing normal physiological activities and pathological processes. Oxidative stress arises while the generation of reactive oxygen species (ROS) overload the neutralizing capacity of intrinsic antioxidants and antioxidative defenses. Intracellular ROS are mainly generated from mitochondrial electron transport chain, NADPH oxidases (NOXs), and xanthine oxidase
[6], as well as, by exogenous stimuli such as electrophiles and ultraviolet light. They are also involved in the regulating of various cellular activities including growth, differentiation, inflammation, infection, ischemia, aging, and disease pathogenesis and progression
[7]. Cellular enzymatic and non-enzymatic antioxidants function as an intrinsic defense to prevent oxidant attack and ameliorate oxidative stress. Enzymatic antioxidants include superoxide dismutases, catalase, peroxiredoxins, and glutathione peroxidase (GPX). The most abundant intracellular non-enzymatic antioxidant is glutathione (GSH)
[6][7][6,7].
Despite the fact that ROS are produced as a byproduct in mitochondria biogenesis and intracellular metabolic activities, they are applied in the transduction of cellular signaling or triggering of intracellular defense. Moderately increased level of ROS promotes the systemic defense by inducing adaptive responses to support cell survival, whereas sustained oxidative stress is associated with many pathological conditions such as cancer, metabolic disorders, and neurodegenerative diseases
[6][7][6,7]. An overload level of ROS can oxidize DNA, RNA, proteins, and lipids, causing irreversible damages and serious oxidative stress that eventually provoke cell death
[6][7][6,7].
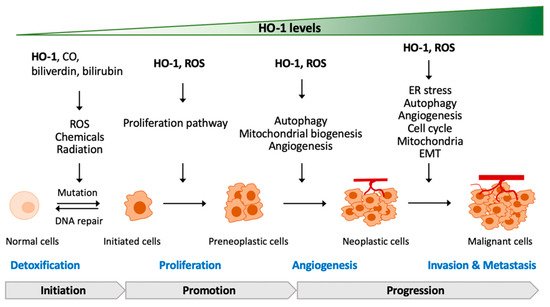
In addition to heme metabolism, HO-1 is induced by a broad range of incitements including oxidants, cytokines, growth factors and hormones, heavy metals, and physical cues (ischemia/reperfusion injury and hypoxia/hyperoxia), especially being highly sensitive to pro-oxidant stimuli, such as ultraviolet, heavy metals, inflammatory cytokines, and iron-containing molecules, that contribute to a regulatory network of cell functions
[8]. Thus, HO-1 is regarded as a pro-oxidant indicator. Several in vivo studies with HO-1-deficiency have demonstrated a cytoprotective effect of HO-1 in human diseases including systemic inflammation, hemolysis, nephritis, asplenia, nephropathy, and vascular endothelial injury
[9][10][9,10]. Results from clinical studies further confirmed that HO-1 protects cells by diminishing oxidative stress and inflammation, and maintaining mitochondrial integrity, thereby promoting cell survival
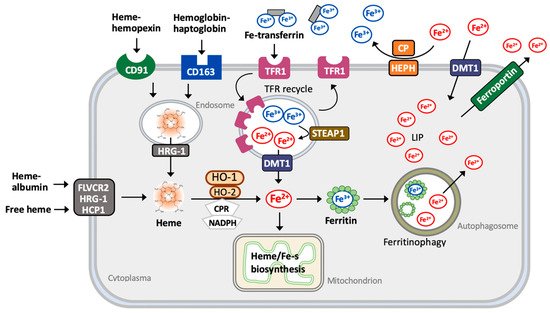
[11]. Due to the manner in regulation of iron metabolism
[12], HO-1 also play a role in mediating ferroptosis, a newly identified iron-dependent cell death
[13][14][15][16][13,14,15,16]. Since heme degradation generates distinctive metabolites including pro-oxidant ferrous iron and anti-oxidant biliverdin
[1][2][1,2], HO-1 apparently possesses a dual role either to protect or deteriorate cancer-cell death
[8][17][18][8,17,18]. The mechanisms of HO-1 in redox homeostasis and how HO-1 interplays with oxidative stress to regulate tumor progression are addressed in the later sections.