 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yusaku Miyamae | + 3664 word(s) | 3664 | 2021-09-13 05:45:37 | | | |
| 2 | Peter Tang | Meta information modification | 3664 | 2021-09-23 09:17:21 | | |
Video Upload Options
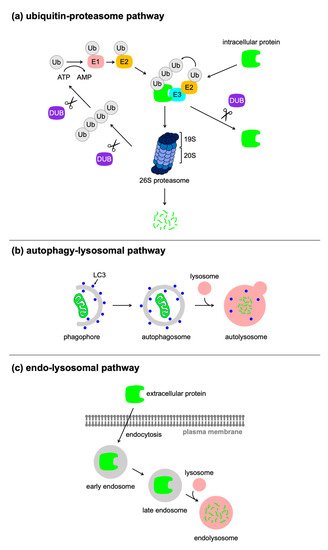
Proteins are fundamental biomolecules of living cells, and their expression levels depend on the balance between the synthesis and degradation. The genetic manipulation of the target protein using CRISPR/Cas9, Cre/loxP, tetracyclin system, and RNA interference, are widely used for the regulation of proteins at the DNA, transcriptional, or mRNA level. Recently, researchers have developed various types of molecular tools for the regulation of protein expression at the post-translational level, which rely on harnessing cellular proteolytic machinery including ubiquitin–proteasome pathway, autophagy-lysosome pathway, and endocytosis. The post-translational control of protein expression using small molecules, antibodies, and light can offer significant advantages regarding speed, tunability, and reversibility. These technologies are expected to be applied to pharmacotherapy and cell therapy, as well as research tools for fundamental biological studies.
1. Introduction
2. Targeted Protein Degradation Using Bifunctional Molecules or Antibodies
2.1. PROTAC
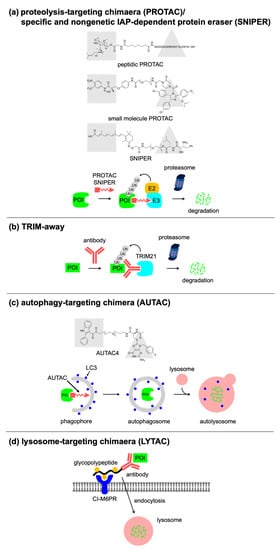
|
Technology |
Proteolytic Pathway |
Feature |
References |
|---|---|---|---|
|
PROTAC/SNIPER |
Ubiquitin–proteasome pathway |
Originally developed bifunctional molecules |
|
|
CLIPTAC |
Ubiquitin–proteasome pathway |
In-cell generation of bifunctional molecule by the biorthogonal reaction |
[24] |
|
opto-PROTAC |
Ubiquitin–proteasome pathway |
Light-inducible switch on PROTAC by photolabile caging group |
[25] |
|
PHOTAC |
Ubiquitin–proteasome pathway |
Light-inducible switch on PROTAC by photo-reversible isomerization of azobenzene moiety |
[26] |
|
AbTAC |
Ubiquitin–proteasome pathway |
Capable of degrading the cell-surface protein |
[27] |
|
Antibody-PROTAC conjugate |
Ubiquitin–proteasome pathway |
Cell-type selective targeting |
[28] |
|
TRIM away |
Ubiquitin–proteasome pathway |
Selective degradation of post-translational modified protein and mutant variants |
|
|
AUTAC |
Autophagy-lysosome pathway |
Capable of targeting cellular organelles |
[31] |
|
ATTEC |
Autophagy-lysosome pathway |
Molecular glue type degrader for targeting abnormal proteins |
|
|
LYTAC |
Endocytosis |
Capable of targeting extracellular and membrane-bound protein |
2.2. Antibody-Based Approaches
2.3. Autophagy/Lysosome-Based Approaches
3. Conditional Control of Protein Stability Using Genetic Manipulation
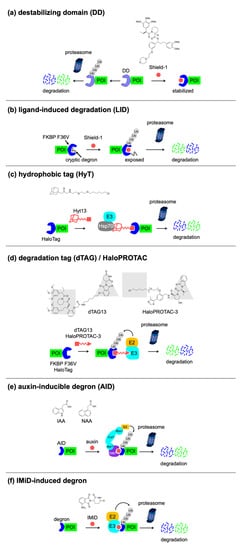
3.1. Small-Molecule-Switchable Degrons
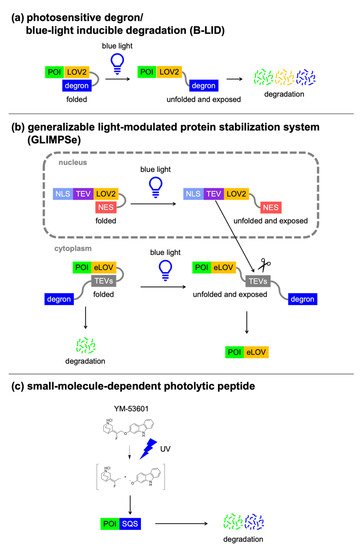
3.2. Light-Switchable Degrons
3.3. Nanobody-Based Degrons
|
Degron Technology |
Tag Size |
Switch |
Number of Component(s) |
Features |
References |
|---|---|---|---|---|---|
|
DD |
FKBP DD (12 kDa), DHFR DD (18 kDa), etc. |
Shield-1 (FKBP DD), TMP (DHFR DD), etc. |
1 |
Drug-induced protein stabilization |
|
|
LID |
FKBP F36V-degron (13 kDa) |
Shield-1 |
1 |
Drug-induced protein degradation |
[47] |
|
HaloPROTAC |
Halo tag (34 kDa) |
HaloPROTAC-3 |
1 |
Drug-induced protein degradation |
[56] |
|
HyT |
Halo tag (34 kDa) |
Hyt13 |
1 |
Drug-induced protein degradation |
[57] |
|
dTAG |
FKBP F36V (12 kDa) |
dTAG13 |
1 |
Drug-induced protein degradation |
[58] |
|
AID |
AID/IAA (25 kDa), mAID (7 kDa) |
Auxin (IAA, NAA) |
2 |
Drug-induced protein degradation |
[59] |
|
IMiD-induced degron |
IKZF3-based degron (6 kDa), SALL4 degron (3 kDa) |
IMiD (thalidomide, pomalidomide, 5-hydroxythalidomide) |
1 |
Drug-induced protein degradation |
|
|
Photosensitive degron/B-LID |
LOV2-degron (20 kDa) |
Blue light |
1 |
Light-induced protein degradation |
|
|
GLIMPSe |
eLOV-TEVs-degron (27 kDa) |
Blue light |
2 |
Light-induced protein stabilization |
[62] |
|
Small-molecule-dependent photolytic peptide |
SQS C-terminal peptide 371–417 (3 kDa) |
YM-53601 & UV |
1 |
Drug- and UV-induced protein degradation, no use of cellular degradation mechanisms |
[63] |
|
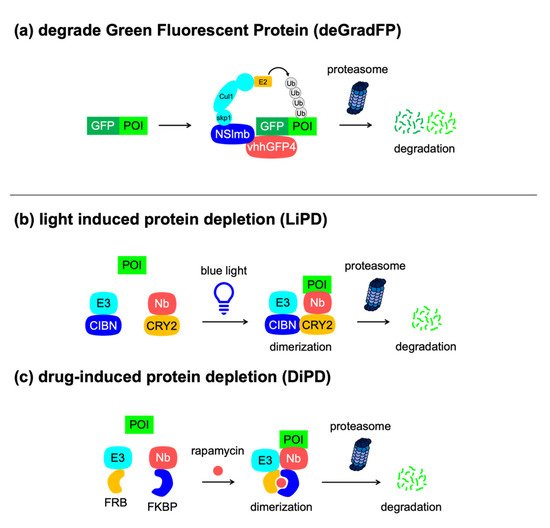
deGradFP |
GFP (25 kDa) |
NA |
2 |
Protein degradation upon expression of the F-box protein, anti-GFP nanobody, no need to attach tags to POIs |
[53] |
|
LiPD |
NA |
Blue light |
2 |
Light-induced protein degradation, no need to attach tags to POIs |
[64] |
|
DiPD |
NA |
Rapamycin |
2 |
Drug-induced protein degradation, no need to attach tags to POIs |
[64] |
|
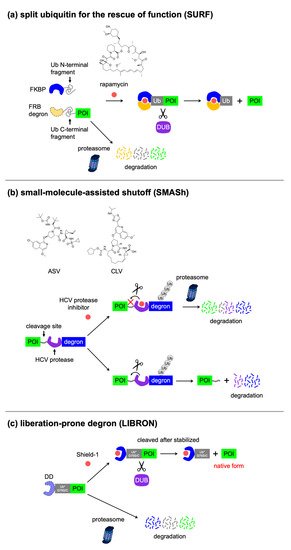
SURF |
3 x FRB degron-Ub-C (58 kDa) |
Rapamycin |
2 |
Drug-induced protein stabilization, tag removal via endogenous protease activity |
[65] |
|
SMASh |
SMASh tag (34 kDa) |
HCV protease inhibitors (ASV, CLV) |
1 |
Drug-induced protein degradation, tag removal via intramolecular protease activity |
[66] |
|
LIBRON |
FKBP DD-Ub (20 kDa) DHFR DD-Ub (27 kDa) |
Shield-1 (FKBP DD-Ub), TMP (DHFR DD-Ub) |
1 |
Drug-induced protein stabilization, tag removal via endogenous protease activity |
[67] |
3.4. Excisable Degrons
References
- Rakhit, R.; Navarro, R.; Wandless, T.J. Chemical biology strategies for posttranslational control of protein function. Chem. Biol. 2014, 21, 1238–1252.
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823.
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826.
- Komor, A.C.; Badran, A.H.; Liu, D.R. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 2017, 168, 20–36.
- Sauer, B.; Henderson, N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc. Natl. Acad. Sci. USA 1988, 85, 5166–5170.
- Sauer, B. Cre/lox: One more step in the taming of the genome. Endocrine 2002, 19, 221–228.
- Le, Y.; Sauer, B. Conditional gene knockout using Cre recombinase. Mol. Biotechnol. 2001, 17, 269–275.
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526.
- Blomen, V.A.; Májek, P.; Jae, L.T.; Bigenzahn, J.W.; Nieuwenhuis, J.; Staring, J.; Sacco, R.; van Diemen, F.R.; Olk, N.; Stukalov, A.; et al. Gene essentiality and synthetic lethality in haploid human cells. Science 2015, 350, 1092–1096.
- Teng, X.; Dayhoff-Brannigan, M.; Cheng, W.C.; Gilbert, C.E.; Sing, C.N.; Diny, N.L.; Wheelan, S.J.; Dunham, M.J.; Boeke, J.D.; Pineda, F.J.; et al. Genome-wide consequences of deleting any single gene. Mol. Cell 2013, 52, 485–494.
- Gossen, M.; Bujard, H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA 1992, 89, 5547–5551.
- Kistner, A.; Gossen, M.; Zimmermann, F.; Jerecic, J.; Ullmer, C.; Lübbert, H.; Bujard, H. Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic mice. Proc. Natl. Acad. Sci. USA 1996, 93, 10933–10938.
- Zhu, Z.; Zheng, T.; Lee, C.G.; Homer, R.J.; Elias, J.A. Tetracycline-controlled transcriptional regulation systems: Advances and application in transgenic animal modeling. Semin. Cell Dev. Biol. 2002, 13, 121–128.
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498.
- Aagaard, L.; Rossi, J.J. RNAi therapeutics: Principles, prospects and challenges. Adv. Drug Deliv. Rev. 2007, 59, 75–86.
- Sacher, R.; Stergiou, L.; Pelkmans, L. Lessons from genetics: Interpreting complex phenotypes in RNAi screens. Curr. Opin. Cell Biol. 2008, 20, 483–489.
- Ravid, T.; Hochstrasser, M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell Biol. 2008, 9, 679–690.
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118.
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Ann. Rev. Biochem. 2017, 86, 159–192.
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559.
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett. 2008, 18, 5904–5908.
- Itoh, Y.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Protein knockdown using methyl bestatin-ligand hybrid molecules: Design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J. Am. Chem. Soc. 2010, 132, 5820–5826.
- Atilaw, Y.; Poongavanam, V.; Svensson Nilsson, C.; Nguyen, D.; Giese, A.; Meibom, D.; Erdelyi, M.; Kihlberg, J. Solution Conformations Shed Light on PROTAC Cell Permeability. ACS Med. Chem. Lett. 2021, 12, 107–114.
- Lebraud, H.; Wright, D.J.; Johnson, C.N.; Heightman, T.D. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2016, 2, 927–934.
- Liu, J.; Chen, H.; Ma, L.; He, Z.; Wang, D.; Liu, Y.; Lin, Q.; Zhang, T.; Gray, N.; Kaniskan, H.; et al. Light-induced control of protein destruction by opto-PROTAC. Sci. Adv. 2020, 6, eaay5154.
- Reynders, M.; Matsuura, B.S.; Bérouti, M.; Simoneschi, D.; Marzio, A.; Pagano, M.; Trauner, D. PHOTACs enable optical control of protein degradation. Sci. Adv. 2020, 6, eaay5064.
- Cotton, A.D.; Nguyen, D.P.; Gramespacher, J.A.; Seiple, I.B.; Wells, J.A. Development of Antibody-Based PROTACs for the Degradation of the Cell-Surface Immune Checkpoint Protein PD-L1. J. Am. Chem. Soc. 2021, 143, 593–598.
- Maneiro, M.A.; Forte, N.; Shchepinova, M.M.; Kounde, C.S.; Chudasama, V.; Baker, J.R.; Tate, E.W. Antibody-PROTAC Conjugates Enable HER2-Dependent Targeted Protein Degradation of BRD4. ACS Chem. Biol. 2020, 15, 1306–1312.
- Clift, D.; McEwan, W.A.; Labzin, L.I.; Konieczny, V.; Mogessie, B.; James, L.C.; Schuh, M. A Method for the Acute and Rapid Degradation of Endogenous Proteins. Cell 2017, 171, 1692–1706.e18.
- Zeng, J.; Santos, A.F.; Mukadam, A.S.; Osswald, M.; Jacques, D.A.; Dickson, C.F.; McLaughlin, S.H.; Johnson, C.M.; Kiss, L.; Luptak, J.; et al. Target-induced clustering activates Trim-Away of pathogens and proteins. Nat. Struct. Mol. Biol. 2021, 28, 278–289.
- Takahashi, D.; Moriyama, J.; Nakamura, T.; Miki, E.; Takahashi, E.; Sato, A.; Akaike, T.; Itto-Nakama, K.; Arimoto, H. AUTACs: Cargo-Specific Degraders Using Selective Autophagy. Mol. Cell 2019, 76, 797–810.e10.
- Li, Z.; Wang, C.; Wang, Z.; Zhu, C.; Li, J.; Sha, T.; Ma, L.; Gao, C.; Yang, Y.; Sun, Y.; et al. Allele-selective lowering of mutant HTT protein by HTT-LC3 linker compounds. Nature 2019, 575, 203–209.
- Li, Z.; Zhu, C.; Ding, Y.; Fei, Y.; Lu, B. ATTEC: A potential new approach to target proteinopathies. Autophagy 2020, 16, 185–187.
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297.
- Ahn, G.; Banik, S.M.; Miller, C.L.; Riley, N.M.; Cochran, J.R.; Bertozzi, C.R. LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation. Nat. Chem. Biol. 2021.
- Varshavsky, A. Naming a targeting signal. Cell 1991, 64, 13–15.
- Banaszynski, L.A.; Chen, L.C.; Maynard-Smith, L.A.; Ooi, A.G.; Wandless, T.J. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 2006, 126, 995–1004.
- Iwamoto, M.; Bjorklund, T.; Lundberg, C.; Kirik, D.; Wandless, T.J. A general chemical method to regulate protein stability in the mammalian central nervous system. Chem. Biol. 2010, 17, 981–988.
- Miyazaki, Y.; Imoto, H.; Chen, L.C.; Wandless, T.J. Destabilizing domains derived from the human estrogen receptor. J. Am. Chem. Soc. 2012, 134, 3942–3945.
- Navarro, R.; Chen, L.C.; Rakhit, R.; Wandless, T.J. A Novel Destabilizing Domain Based on a Small-Molecule Dependent Fluorophore. ACS Chem. Biol. 2016, 11, 2101–2104.
- Banaszynski, L.A.; Sellmyer, M.A.; Contag, C.H.; Wandless, T.J.; Thorne, S.H. Chemical control of protein stability and function in living mice. Nat. Med. 2008, 14, 1123–1127.
- Armstrong, C.M.; Goldberg, D.E. An FKBP destabilization domain modulates protein levels in Plasmodium falciparum. Nat. Methods 2007, 4, 1007–1009.
- Herm-Gotz, A.; Agop-Nersesian, C.; Munter, S.; Grimley, J.S.; Wandless, T.J.; Frischknecht, F.; Meissner, M. Rapid control of protein level in the apicomplexan Toxoplasma gondii. Nat. Methods 2007, 4, 1003–1005.
- Cho, U.; Zimmerman, S.M.; Chen, L.C.; Owen, E.; Kim, J.V.; Kim, S.K.; Wandless, T.J. Rapid and tunable control of protein stability in Caenorhabditis elegans using a small molecule. PLoS ONE 2013, 8, e72393.
- Sethi, S.; Wang, J.W. A versatile genetic tool for post-translational control of gene expression in Drosophila melanogaster. eLife 2017, 6, e30327.
- Ramadurgum, P.; Daniel, S.; Hulleman, J.D. Protocol for In Vivo Evaluation and Use of Destabilizing Domains in the Eye, Liver, and Beyond. STAR Protoc. 2020, 1, 100094.
- Bonger, K.M.; Chen, L.C.; Liu, C.W.; Wandless, T.J. Small-molecule displacement of a cryptic degron causes conditional protein degradation. Nat. Chem. Biol. 2011, 7, 531–537.
- Jariel-Encontre, I.; Bossis, G.; Piechaczyk, M. Ubiquitin-independent degradation of proteins by the proteasome. Biochim. Biophys. Acta 2008, 1786, 153–177.
- Renicke, C.; Schuster, D.; Usherenko, S.; Essen, L.O.; Taxis, C. A LOV2 domain-based optogenetic tool to control protein degradation and cellular function. Chem. Biol. 2013, 20, 619–626.
- Bonger, K.M.; Rakhit, R.; Payumo, A.Y.; Chen, J.K.; Wandless, T.J. General method for regulating protein stability with light. ACS Chem. Biol. 2014, 9, 111–115.
- Rothbauer, U.; Zolghadr, K.; Tillib, S.; Nowak, D.; Schermelleh, L.; Gahl, A.; Backmann, N.; Conrath, K.; Muyldermans, S.; Cardoso, M.C.; et al. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat. Methods 2006, 3, 887–889.
- Hansen, S.; Stuber, J.C.; Ernst, P.; Koch, A.; Bojar, D.; Batyuk, A.; Pluckthun, A. Design and applications of a clamp for Green Fluorescent Protein with picomolar affinity. Sci. Rep. 2017, 7, 16292.
- Caussinus, E.; Kanca, O.; Affolter, M. Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat. Struct. Mol. Biol. 2011, 19, 117–121.
- Ludwicki, M.B.; Li, J.; Stephens, E.A.; Roberts, R.W.; Koide, S.; Hammond, P.T.; DeLisa, M.P. Broad-Spectrum Proteome Editing with an Engineered Bacterial Ubiquitin Ligase Mimic. ACS Cent. Sci. 2019, 5, 852–866.
- Daniel, K.; Icha, J.; Horenburg, C.; Muller, D.; Norden, C.; Mansfeld, J. Conditional control of fluorescent protein degradation by an auxin-dependent nanobody. Nat. Commun. 2018, 9, 3297.
- Buckley, D.L.; Raina, K.; Darricarrere, N.; Hines, J.; Gustafson, J.L.; Smith, I.E.; Miah, A.H.; Harling, J.D.; Crews, C.M. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 2015, 10, 1831–1837.
- Neklesa, T.K.; Tae, H.S.; Schneekloth, A.R.; Stulberg, M.J.; Corson, T.W.; Sundberg, T.B.; Raina, K.; Holley, S.A.; Crews, C.M. Small-molecule hydrophobic tagging-induced degradation of HaloTag fusion proteins. Nat. Chem. Biol. 2011, 7, 538–543.
- Nabet, B.; Roberts, J.M.; Buckley, D.L.; Paulk, J.; Dastjerdi, S.; Yang, A.; Leggett, A.L.; Erb, M.A.; Lawlor, M.A.; Souza, A.; et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018, 14, 431–441.
- Nishimura, K.; Fukagawa, T.; Takisawa, H.; Kakimoto, T.; Kanemaki, M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods 2009, 6, 917–922.
- Koduri, V.; McBrayer, S.K.; Liberzon, E.; Wang, A.C.; Briggs, K.J.; Cho, H.; Kaelin, W.G., Jr. Peptidic degron for IMiD-induced degradation of heterologous proteins. Proc. Natl. Acad. Sci. USA 2019, 116, 2539–2544.
- Yamanaka, S.; Shoya, Y.; Matsuoka, S.; Nishida-Fukuda, H.; Shibata, N.; Sawasaki, T. An IMiD-induced SALL4 degron system for selective degradation of target proteins. Commun. Biol. 2020, 3, 515.
- Mondal, P.; Krishnamurthy, V.V.; Sharum, S.R.; Haack, N.; Zhou, H.; Cheng, J.; Yang, J.; Zhang, K. Repurposing Protein Degradation for Optogenetic Modulation of Protein Activities. ACS Synth. Biol. 2019, 8, 2585–2592.
- Takemoto, Y.; Mao, D.; Punzalan, L.L.; Gotze, S.; Sato, S.I.; Uesugi, M. Discovery of a Small-Molecule-Dependent Photolytic Peptide. J. Am. Chem. Soc. 2020, 142, 1142–1146.
- Deng, W.; Bates, J.A.; Wei, H.; Bartoschek, M.D.; Conradt, B.; Leonhardt, H. Tunable light and drug induced depletion of target proteins. Nat. Commun. 2020, 11, 304.
- Pratt, M.R.; Schwartz, E.C.; Muir, T.W. Small-molecule-mediated rescue of protein function by an inducible proteolytic shunt. Proc. Natl. Acad. Sci. USA 2007, 104, 11209–11214.
- Chung, H.K.; Jacobs, C.L.; Huo, Y.; Yang, J.; Krumm, S.A.; Plemper, R.K.; Tsien, R.Y.; Lin, M.Z. Tunable and reversible drug control of protein production via a self-excising degron. Nat. Chem. Biol. 2015, 11, 713–720.
- Miyamae, Y.; Chen, L.C.; Utsugi, Y.; Farrants, H.; Wandless, T.J. A Method for Conditional Regulation of Protein Stability in Native or Near-Native Form. Cell Chem. Biol. 2020, 27, 1573–1581.e3.


