 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mohammed Abdullah Alshawsh | + 2851 word(s) | 2851 | 2021-09-14 05:20:50 | | | |
| 2 | Bruce Ren | -13 word(s) | 2838 | 2021-09-15 03:17:59 | | |
Video Upload Options
The Hedgehog (Hh)-glioma-associated oncogene homolog (GLI) signaling pathway is highly conserved among mammals, with crucial roles in regulating embryonic development as well as in cancer initiation and progression. The GLI transcription factors (GLI1, GLI2, and GLI3) are effectors of the Hh pathway and are regulated via Smoothened (SMO)-dependent and SMO-independent mechanisms. The SMO-dependent route involves the common Hh-PTCH-SMO axis, and mutations or transcriptional and epigenetic dysregulation at these levels lead to the constitutive activation of GLI transcription factors. Conversely, the SMO-independent route involves the SMO bypass regulation of GLI transcription factors by external signaling pathways and their interacting proteins or by epigenetic and transcriptional regulation of GLI transcription factors expression. Both routes of GLI activation, when dysregulated, have been heavily implicated in tumorigenesis of many known cancers, making them important targets for cancer treatment.
1. Introduction
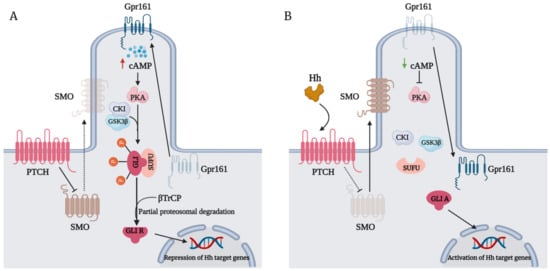
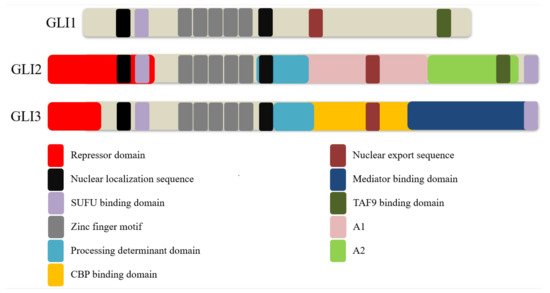
2. GLI Proteins and Their Domains
References
- Nüsslein-volhard, C.; Wieschaus, E. Mutations Affecting Segment Number and Polarity in Drosophila. Nature 1980, 287, 795–801.
- Carballo, G.B.; Honorato, J.R.; De Lopes, G.P.F.; de Sampaio, E.; Spohr, T.C.L. A Highlight on Sonic Hedgehog Pathway. Cell Commun. Signal. 2018, 16, 11.
- Mastronardi, F.G.; Dimitroulakos, J.; Kamel-Reid, S.; Manoukian, A.S. Co-Localization of Patched and Activated Sonic Hedgehog to Lysosomes in Neurons. Neuroreport 2000, 11, 581–585.
- Rohatgi, R.; Milenkovic, L.; Corcoran, R.B.; Scott, M.P. Hedgehog Signal Transduction by Smoothened: Pharmacologic Evidence for a 2-Step Activation Process. Proc. Natl. Acad. Sci. USA 2009, 106, 3196–3201.
- Hsu, S.H.C.; Zhang, X.; Yu, C.; Li, Z.J.; Wunder, J.S.; Hui, C.C.; Alman, B.A. Kif7 Promotes Hedgehog Signaling in Growth Plate Chondrocytes by Restricting the Inhibitory Function of Sufu. Development 2011, 138, 3791–3801.
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The Role of the Hedgehog Signaling Pathway in Cancer: A Comprehensive Review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20.
- Gonnissen, A.; Isebaert, S.; Haustermans, K. Targeting the Hedgehog Signaling Pathway in Cancer: Beyond Smoothened. Oncotarget 2015, 6, 13899–13913.
- Jia, J.; Zhang, L.; Zhang, Q.; Tong, C.; Wang, B.; Hou, F.; Amanai, K.; Jiang, J. Phosphorylation by Double-Time/CKIε and CKIα Targets Cubitus Interruptus for Slimb/β-TRCP-Mediated Proteolytic Processing. Dev. Cell 2005, 9, 819–830.
- Tschaikner, P.; Enzler, F.; Torres-Quesada, O.; Aanstad, P.; Stefan, E. Hedgehog and Gpr161: Regulating CAMP Signaling in the Primary Cilium. Cells 2020, 9, 118.
- Niewiadomski, P.; Kong, J.H.; Ahrends, R.; Ma, Y.; Humke, E.W.; Khan, S.; Teruel, M.N.; Novitch, B.G.; Rohatgi, R. Gli Protein Activity Is Controlled by Multisite Phosphorylation in Vertebrate Hedgehog Signaling. Cell Rep. 2014, 6, 168–181.
- Price, M.A.; Kalderon, D. Proteolysis of the Hedgehog Signaling Effector Cubitus Interruptus Requires Phosphorylation by Glycogen Synthase Kinase 3 and Casein Kinase 1. Cell 2002, 108, 823–835.
- Shafique, S.; Rashid, S. Structural Basis of ΒTrCP1-Associated GLI3 Processing. Sci. Rep. 2019, 9, 6865.
- Pan, Y.; Wang, C.; Wang, B. Phosphorylation of Gli2 by Protein Kinase A Is Required for Gli2 Processing and Degradation and the Sonic Hedgehog-Regulated Mouse Development. Dev. Biol. 2009, 326, 177–189.
- Sabol, M.; Trnski, D.; Musani, V.; Ozretić, P.; Levanat, S. Role of GLI Transcription Factors in Pathogenesis and Their Potential as New Therapeutic Targets. Int. J. Mol. Sci. 2018, 19, 2562.
- Zubčić, V.; Rinčić, N.; Kurtović, M.; Trnski, D.; Musani, V.; Ozretić, P.; Levanat, S.; Leović, D.; Sabol, M. GANT61 and Lithium Chloride Inhibit the Growth of Head and Neck Cancer Cell Lines Through the Regulation of GLI3 Processing by GSK3β. Int. J. Mol. Sci. 2020, 21, 6410.
- Miyazaki, Y.; Matsubara, S.; Ding, Q.; Tsukasa, K.; Yoshimitsu, M.; Kosai, K.i.; Takao, S. Efficient Elimination of Pancreatic Cancer Stem Cells by Hedgehog/GLI Inhibitor GANT61 in Combination with MTOR Inhibition. Mol. Cancer 2016, 15, 49.
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-Canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors beyond Smoothened. Front. Genet. 2019, 10, 556.
- Zhu, H.; Lo, H.-W. The Human Glioma-Associated Oncogene Homolog 1 (GLI1) Family of Transcription Factors in Gene Regulation and Diseases. Curr. Genomics 2010, 11, 238–245.
- Niewiadomski, P.; Niedziółka, S.M.; Markiewicz, Ł.; Uśpieński, T.; Baran, B.; Chojnowska, K. Gli Proteins: Regulation in Development and Cancer. Cells 2019, 8, 147.
- Pan, Y.; Wang, B. A Novel Protein-Processing Domain in Gli2 and Gli3 Differentially Blocks Complete Protein Degradation by the Proteasome. J. Biol. Chem. 2007, 282, 10846–10852.
- Kinzler, K.W.; Vogelstein, B. The GLI Gene Encodes a Nuclear Protein Which Binds Specific Sequences in the Human Genome. Mol. Cell. Biol. 1990, 10, 634.
- Pavletich, N.; Pabo, C. Crystal Structure of a Five-Finger GLI-DNA Complex: New Perspectives on Zinc Fingers. Science 1993, 261, 1701–1707.
- Hatayama, M.; Aruga, J. Gli Protein Nuclear Localization Signal. Vitam. Horm. 2012, 88, 73–89.
- Szczepny, A.; Wagstaff, K.M.; Dias, M.; Gajewska, K.; Wang, C.; Davies, R.G.; Kaur, G.; Ly-Huynh, J.; Loveland, K.L.; Jans, D.A. Overlapping Binding Sites for Importin Β1 and Suppressor of Fused (SuFu) on Glioma-Associated Oncogene Homologue 1 (Gli1) Regulate Its Nuclear Localization. Biochem. J. 2014, 461, 469–476.
- Torrado, B.; Graña, M.; Badano, J.L.; Irigoín, F. Ciliary Entry of the Hedgehog Transcriptional Activator Gli2 Is Mediated by the Nuclear Import Machinery but Differs from Nuclear Transport in Being Imp-α/Β1-Independent. PLoS ONE 2016, 11, e0162033.
- Barnfield, P.C.; Zhang, X.; Thanabalasingham, V.; Yoshida, M.; Hui, C. Negative Regulation of Gli1 and Gli2 Activator Function by Suppressor of Fused through Multiple Mechanisms. Differentiation 2005, 73, 397–405.
- Sheng, T.; Chi, S.; Zhang, X.; Xie, J. Regulation of Gli1 Localization by the CAMP/Protein Kinase A Signaling Axis through a Site Near the Nuclear Localization Signal. J. Biol. Chem. 2006, 281, 9–12.
- Shi, Q.; Han, Y.; Jiang, J. Suppressor of Fused Impedes Ci/Gli Nuclear Import by Opposing Trn/Kapb2 in Hedgehog Signaling. J. Cell Sci. 2014, 127, 1092–1103.
- Han, Y.; Xiong, Y.; Shi, X.; Wu, J.; Zhao, Y.; Jiang, J. Regulation of Gli Ciliary Localization and Hedgehog Signaling by the PY-NLS/Karyopherin-Β2 Nuclear Import System. PLOS Biol. 2017, 15, e2002063.
- Kogerman, P.; Grimm, T.; Kogerman, L.; Krause, D.; Undén, A.B.; Sandstedt, B.; Toftgård, R.; Zaphiropoulos, P.G. Mammalian Suppressor-of-Fused Modulates Nuclear-Cytoplasmic Shuttling of GLI-1. Nat. Cell Biol. 1999, 1, 312–319.
- Han, Y.; Shi, Q.; Jiang, J. Multisite Interaction with Sufu Regulates Ci/Gli Activity through Distinct Mechanisms in Hh Signal Transduction. Proc. Natl. Acad. Sci. USA 2015, 112, 6383–6388.
- Zhang, Z.; Shen, L.; Law, K.; Zhang, Z.; Liu, X.; Hua, H.; Li, S.; Huang, H.; Yue, S.; Hui, C.; et al. Suppressor of Fused Chaperones Gli Proteins To Generate Transcriptional Responses to Sonic Hedgehog Signaling. Mol. Cell. Biol. 2017, 37, e00421-16.
- Akimaru, H.; Chen, Y.; Dai, P.; Hou, D.-X.; Nonaka, M.; Smolik, S.M.; Armstrong, S.; Goodman, R.H.; Ishii, S. Drosophila CBP Is a Co-Activator of Cubitus Interruptus in Hedgehog Signalling. Nature 1997, 386, 735–738.
- Hughes, D.C.; Allen, J.; Morley, G.; Sutherland, K.; Ahmed, W.; Prosser, J.; Lettice, L.; Allan, G.; Mattei, M.G.; Farrall, M.; et al. Cloning and Sequencing of the Mouse Gli2 Gene: Localization to the Dominant Hemimelia Critical Region. Genomics 1997, 39, 205–215.
- Dai, P.; Akimaru, H.; Tanaka, Y.; Maekawa, T.; Nakafuku, M.; Ishii, S. Sonic Hedgehog-Induced Activation of the Gli1Promoter Is Mediated by GLI3. J. Biol. Chem. 1999, 274, 8143–8152.
- Zhou, H.; Kim, S.; Ishii, S.; Boyer, T.G. Mediator Modulates Gli3-Dependent Sonic Hedgehog Signaling. Mol. Cell. Biol. 2006, 26, 8667.
- Yoon, J.W.; Liu, C.Z.; Yang, J.T.; Swart, R.; Iannaccone, P.; Walterhouse, D. GLI Activates Transcription through a Herpes Simplex Viral Protein 16-Like Activation Domain. J. Biol. Chem. 1998, 273, 3496–3501.
- Yoon, J.W.; Lamm, M.; Iannaccone, S.; Higashiyama, N.; Leong, K.F.; Iannaccone, P.; Walterhouse, D. P53 Modulates The Activity Of The GLI1 Oncogene Through Interactions With The Shared Coactivator TAF9. DNA Repair 2015, 34, 9.
- Bosco-Clément, G.; Zhang, F.; Chen, Z.; Zhou, H.M.; Li, H.; Mikami, I.; Hirata, T.; Yagui-Beltran, A.; Lui, N.; Do, H.T.; et al. Targeting Gli Transcription Activation by Small Molecule Suppresses Tumor Growth. Oncogene 2014, 33, 2087–2097.
- Dai, P.; Shinagawa, T.; Nomura, T.; Harada, J.; Kaul, S.C.; Wadhwa, R.; Khan, M.M.; Akimaru, H.; Sasaki, H.; Colmenares, C.; et al. Ski Is Involved in Transcriptional Regulation by the Repressor and Full-Length Forms of Gli3. Genes Dev. 2002, 16, 2843.
- Cheng, S.Y.; Bishop, J.M. Suppressor of Fused Represses Gli-Mediated Transcription by Recruiting the SAP18-MSin3 Corepressor Complex. Proc. Natl. Acad. Sci. USA 2002, 99, 5442–5447.


