1000/1000
Hot
Most Recent
 +1 point
+1 point
The immunological synapse (IS) is an intercellular communication platform, organized at the contact site of two adjacent cells, where at least one is an immune cell. Functional IS formation is fundamental for the modulation of the most relevant immune system activities, such as T cell activation by antigen presenting cells. Extensive evidence suggests that connexins, in particular connexin-43 (Cx43) hemichannels and/or gap junctions (GJ), regulate signaling events in different types of IS. Although the underlying mechanisms are not fully understood, the current evidence suggests that Cx43 channels could act as facilitators for calcium ions, cyclic adenosine monophosphate, and/or adenosine triphosphate uptake and/or release at the interface of interacting cells. These second messengers have relevant roles in the IS signaling during dendritic cell (DC)-mediated T and NK cell activation, regulatory T cell-mediated immune suppression, and cytotoxic T lymphocyte or NK cell-mediated target tumor cell killing.
The immunological synapse (IS) is a specialized contact area formed between two adjacent cells, where at least one of them is an immune cell. This cell contact structure is characterized by a close apposition of an immune cell membrane with the membrane of an adjacent cell, induced by adaptive or innate immune recognition, intercellular adhesion, stability and polarized signaling.
The formation of a functional IS is fundamental for the modulation of most relevant immune system activities, such as the priming and activation of T (cytotoxic CD8+ and helper CD4+) and natural killer (NK) cells by professional antigen presenting cells (APCs), like dendritic cells (DC), macrophages, and B cells [1][2]; killing of target (infected or cancer) cells by NK cells and cytotoxic T lymphocytes (CTL), via the formation of a cytotoxic IS (CIS) [3]; phagocytosis of microbes by myeloid phagocytes [4]; inflammatory responses mediated by mast cells via an antibody-dependent degranulatory synapse [5]; antigen extraction, processing and presentation by B cells [6]; and regulatory T cell (Treg)-mediated immune suppression [7].
Regardless of the type of interacting immune cell, a mature IS comprises highly ordered and plastic signaling platforms that integrate signals and coordinates molecular interactions leading to appropriate immune responses [8]. These signaling platforms are organized in at least three concentric regions called supramolecular activation clusters (SMAC): the central, the peripheral and the distal SMAC (cSMAC, pSMAC and dSMAC, respectively) [9][10]. These organized structures are more characteristic of T and B cell IS, but some of these molecular organizations are also found in the CIS from NK cells [11]. In general, the cSMAC, a molecular platform that mediates both proximal signaling events and active secretion, is organized as a cluster of T cell receptor (TCR), B cell receptor (BCR) or activating/inhibitory NK cell receptors, associated signaling molecules, co-stimulatory receptor/ligands, and a secretory domain. The pSMAC includes adhesion molecule interactions, like lymphocyte function-associated antigen-1 (LFA-1)/intercellular adhesion molecule-I (ICAM-1), which promote the stable adhesion of interacting cells; whereas a ring of filamentous actin (F-actin), which exerts mechanical forces required for IS activity, is generally accumulated at the dSMAC (Figure 1) [9][10][12].
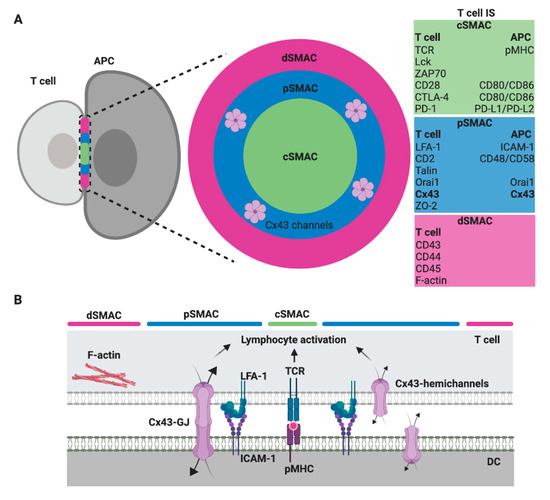
Figure 1. Scheme of a T cell immunological synapse (IS) and localization of Cx43 formed gap junctions (GJ) in the SMAC. (A) A face on view of the IS with the characteristic SMAC patterns, including the cSMAC (green), the pSMAC ring surrounding the cSMAC (blue) and the distal region to the synapse outside the pSMAC (dSMAC, red), as well as the molecules/ligand that are found enriched within. The evidence suggests that gap junction (GJ) channels formed by Cx43 (Cx43-GJ), as well as Cx43 hemichannels, are located in the pSMAC region [13]. (B) A profile view showing a selection of key ligand pairs and Cx43 channels (GJ and hemichannels) that are involved in DC-mediated T cell activation.
Gap junctions (GJ) are clusters of intercellular channels found at the plasma membrane of interacting cells that allow its direct communication. Each GJ is formed by two connexons, which are hexameric hemichannels of connexin (Cx) proteins inserted into the plasma membrane of the cells, each one provided by each of the two contacting cells [14]. These Cx-formed hemichannels can also work as uncoupled channels, allowing the transfer of chemical information from the cytoplasm to the extracellular milieu, and vice versa. Once functional Cx-channels are established, they allow the bidirectional transfer of small molecules (up to ≈1.4 nm) of varied nature, including adenosine triphosphate (ATP), cyclic adenosine monophosphate (cAMP), inositol triphosphate (IP3), calcium, small peptides (including antigens), and microRNAs [15]. There are 20 Cx members in mice and 21 in humans, and the different isoforms determine channel properties. Cxs are generally expressed in a tissue-specific manner, with the exception of Cx43, that is expressed almost ubiquitously and is the main Cx member expressed in the immune system cells [16]. Practically all the immune cells and their hematopoietic precursors express Cx43, and nowadays, its participation in the modulation of different aspects of immune responses is well recognized, as we and other groups recently reviewed [17][18]. Other Cx isoforms like Cx45, Cx40, Cx37, Cx30.3, and Cx26 have also been implicated in inflammatory or other immunological events [19][20][21][22]. However, until now, a majority of accumulated data in immunological literature refers to Cx43, probably due to a higher development of molecular tools, such as specific antibodies and inhibitors.
Eighteen years have passed since Oviedo-Orta and Evans suggested, for the first time, a role for GJ and Cx as potential contributors to the IS signaling [23].
An efficient T cell-mediated adaptive immunity relies on the ability of these cells to differentiate and to make a concerted response. Therefore, an immune response requires that specific T cells find a cognate antigenic peptide complexed to major histocompatibility complex (MHC) molecules (pMHC) on the surface of antigen presenting cells (APCs), and receive appropriate signals to differentiate into effector (Teff), regulatory (Treg) or memory subsets. These fundamental signals are sensed by the T cell during the establishment of an immunological synapse (IS) with the dendritic cell (DC), and are typically categorized in three different types: signal 1, which is supported by antigen recognition (TCR-pMHC); signal 2, corresponding to the co-stimulation given by the interaction of CD28-CD80/CD86, CD40-CD40L, ICOS-ICOSL, among others; and signal 3, which is an instructive cytokine-driven signal [24] (Figure 1). Additional upstream signaling events are also fundamental for T cell priming and the polarization of T cell-mediated responses. These signals are related to how DCs were activated and their migration induced by pattern recognition receptors ligation by pathogen-associated molecular patterns and/or danger-associated molecular patterns. These molecular events have been designed as the signal 0, and provoke the start of an immune response [25]. Nevertheless, even more upstream, the activation of cell death pathways on infected or tumor cells, from where the cognate antigens derived, has been proposed as an initiating immunological event, resulting in the generation of the signal −1 [26]. Moreover, additional signaling pathways downstream of TCR activation modulate the strength and nature of the DC-mediated T cell activation. We termed one of these events as “intercellular coupling by channels” and we consider it as signal 4 (Figure 2). As we described below, these mechanisms are of extreme relevance in the signaling amplification induced by pMHC-TCR interactions.
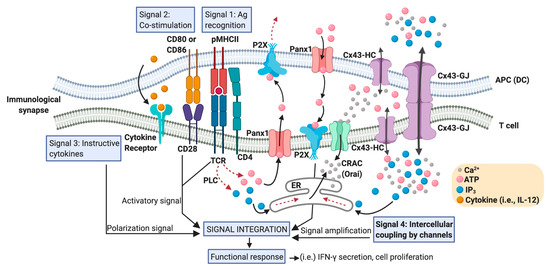
Figure 2. Positioning Cx43 in the intercellular coupling by channels in the DC-T cell IS. An IS between an APC (particularly a DC) and a T cell that effectively promotes T cell activation contains a large number of signaling molecules and signaling events that converge in order to generate T cell functional responses. These signaling events include the activation signal produced by TCR-pMHC ligation (antigen recognition: signal 1), plus co-stimulatory receptor engagement (signal 2), and a polarization signal yielded by instructive cytokines released by the APC (signal 3). Calcium release activated calcium channels (CRAC), such as Orai; hemichannels such as those formed by Panx1 and Cx43; and Cx43 formed gap junctions (Cx43-GJs), are involved in the Ca2+- and ATP- (via purinergic P2X receptors) mediated signaling amplification required for antigen-dependent T cell activation. Although Ca2+ can diffuse through Cx43 channels, the current consensus is that inositol triphosphate (IP3) is the critical intercellular signal that modulates Ca2+ release from internal cellular stores, like endoplasmic reticulum (ER), and it is responsible for GJ-mediated Ca2+ waves in other cell models. An increase in the levels of IP3 in T cells could be produced by the convergence of those generated by phospholipase C (PLC) activation and those acquired from DC via Cx43 (or other Cx isoform) channel-mediated communication. We called these signaling events ‘intercellular coupling by channels’ and considered them as signal 4 in a DC-T cell IS. In response to TCR and CD28 stimulation, Panx1, purinergic P2X receptors, CRAC, and Cx43 translocate to the IS. ATP released through hemichannels promotes autocrine/paracrine signaling via the P2X receptors and the entry of Ca2+ into T cell. Curved arrows indicate movement of molecules; dotted arrows indicate a signaling pathway; straight arrows connect concepts.
Recent evidences suggest that purinergic signaling, together with the global and local accumulation of Ca2+ at the IS proximity, serve as the signal amplification mechanisms needed for T cell activation driven by antigen recognition [27][28][29]. The intercellular coupling by Cx- and pannexin (Panx)-formed channels could have a fundamental role in these processes. Panxs are a family of proteins similar to Cxs, consisting of three members, Panx 1, 2, and 3 [30]. Panxs can form plasma membrane hemichannels, but unlike Cxs, do not produce canonical GJs [31].
T cells and DCs express many members of the purinergic receptor families (P2X, P2Y and P1) [27], and can release ATP in response to various extracellular stimuli, including antigen-specific interactions [32][33][34][35][36]. Upon TCR engagement with anti-CD3/CD28 antibody-coated beads, T cells use Panx1- and Cx-formed hemichannels to release ATP, resulting in the subsequent activation of purinergic receptors P2Xs, thus facilitating both the influx of extracellular Ca2+ and the expression of effector cytokines [32][35][36]. Interestingly, TCR stimulation with anti-CD3/CD28 antibody-coated beads also results in the polarization of P2X1, P2X4 receptors, calcium channels (STIM1 and Orai1), and Panx1 hemichannels to the IS [33]. The inhibition of hemichannels with Panx1-specific mimetic peptides or by carbenoxolone, which is a chemical inhibitor of both Panx- and Cx-formed channels, suppresses TCR-induced ATP release, Ca2+ entry and T cell activation [33], indicating that purinergic signaling plays an active role at the IS in DC-mediated T cell activation.
Upon activation, Cx43 expression, both at mRNA and protein level, increases in DCs and T cells [37][38][39][40][41], suggesting that these cells acquire the capacity to interact with each other through Cx43 channels in order to modulate adaptive immune responses. In this sense, Elgueta and coworkers showed the formation of functional GJ between murine splenic or bone marrow-derived DCs with CD4+ and CD8+ T cells [40]. In their studies, they used two different TCR-transgenic antigenic models: pigeon cytochrome-c peptide presented in the context of I-Ek molecules, and ovalbumin peptides presented in H2-Kb and I-Ab molecules. The formation of GJ-mediated coupling between DC and T cells was sensitive to chemical inhibitors of GJs as oleamide and, more importantly, to Cx43-mimetic blocking peptides [42]. The inhibition of GJs reduced DC-mediated T cell activation, reflected by lower T cell proliferation, CD69 expression and IL-2 secretion. Interestingly, the authors demonstrated that Cx43-GJ blockers did not affect the polyclonal activation of CD4+ T cells induced with soluble anti-CD3/anti-CD28 antibodies in the absence of DCs, indicating that the inhibition of Cx43 channels inhibit T cell activation by directly interfering with GJ assembly between DCs and T cells. Of note, this last experiment differs from those described in the previous paragraph (references 31, 32, 34), in that TCR activation was performed with soluble instead of bead-coated-antibodies. Whereas beads promote the formation of an IS-like structure in the T cells, soluble antibodies do not [43], thus reinforcing the idea of the role of synaptic Cx43 channels in the T cell activation.
In this context, we reported that both hemichannels and GJ formed by Cx43 (Cx43-GJ), polarize to the DC-T cell IS in an antigen- and actin cytoskeleton-dependent manner in murine and human cell models [44]. Indeed, we described that Cx43 channels accumulate preferentially in the pSMAC, colocalizing with LFA-1 molecules. Using Cx43-specific inhibitors, we also showed that Cx43-mediated intercellular communication between DC and T cells is bidirectional [44]. Although the nature of the molecules passing by Cx43 channels in the DC-T cell IS was not fully determined, the silencing of Cx43 expression (either on DCs or T cells) or the inhibition of Cx43-GJ docking, strongly impaired both IFN-γ secretion and the increase of intracellular Ca2+ in the T cells interacting with antigen-loaded DCs [44]. These results strongly suggest that IS-located Cx43 channels participate, directly (as the Orai1 calcium channels) or indirectly, via the release of ATP (as Panx1 hemichannels) and/or through the uptake of IP3 from DC (as described in other cell models [45]), in the Ca2+ elevation required for signaling amplification and T cell activation (Figure 2).
In addition to its role in DC-mediated activation of Teff cells, Cx43 channels also participate in the modulation of the regulatory activity of CD4+ Tregs. Treg cells, which are fundamental players in immune homeostasis and protection against autoimmunity, mediate their suppressive action by acting directly on Teff cells or DCs, through cell contact-dependent and -independent mechanisms. Among the cell contact-dependent mechanisms, the Cx43-GJ-mediated transfer of cAMP is one of the most relevant. Treg and Teff cells showed differential expression and activation of enzymes that regulate intracellular cAMP levels, which translates in Tregs having higher levels of cAMP than Teff cells [46][47]. In 2007, Bopp and coworkers showed that naturally occurring Tregs inhibit CD4+ Teff proliferation, CD69 expression and IL-2 production through the transfer of cAMP via Cx43-GJ-mediated intercellular communications, in vitro as well as in vivo [47]. This second messenger, once inside the cell, triggers different downstream signaling cascades, leading to its immune regulation [48]. In agreement with data showing that DCs are the primary target of Treg cell-mediated suppression [49], the amount of cAMP transferred via Cx43-GJs from Tregs to DCs is significantly higher than that transferred to Teff cells [50]. Indeed, the immune regulation of DCs by cAMP transferred from Tregs via Cx43-GJs has been proposed as a mechanism for controlling graft-versus-host disease after allogeneic hematopoietic stem cell transplantation [50]. Additionally, it has been shown that the reduced suppressive potency of Treg cells of non-obese diabetic (NOD) mice is due to its impaired Cx43 expression and lower capacity to form GJ channels [51]. Indeed, Cx43 overexpression or the strengthening of Cx43-GJ-mediated intercellular communications using the α-Cx carboxyl-terminal synthetic Cx43 mimic peptide 1 (αCT-1), which enhances GJ aggregation by disrupting the interaction between Cx43 and zonula occludens (ZO)-1 [52], increases the suppressive properties of these NOD-derived Treg cells [51]. GJ-mediated coupling between Treg cells and lymph node-resident DCs has also been observed during the induction of tolerance to low doses of allergens in mice [53]. Moreover, Treg cells also abrogate the de novo induction of hapten-specific CD8+ T cell-driven immune reactions, by interfering with T cell stimulatory activity of DCs through Cx43-GJ intercellular communication [54]. Importantly, Cx43-GJ blockage between Tregs and Teff cells abolished Treg cell-mediated suppression of human immunodeficiency virus replication, indicating that Cx43 channels also have an important impact in the outcome of this viral infection [55].


