Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Giampaolo Vetta | + 2012 word(s) | 2012 | 2021-08-23 05:01:15 | | | |
| 2 | Bruce Ren | -13 word(s) | 1999 | 2021-09-10 04:25:19 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Vetta, G. Flecainide in Ventricular Arrhythmias. Encyclopedia. Available online: https://encyclopedia.pub/entry/14050 (accessed on 07 February 2026).
Vetta G. Flecainide in Ventricular Arrhythmias. Encyclopedia. Available at: https://encyclopedia.pub/entry/14050. Accessed February 07, 2026.
Vetta, Giampaolo. "Flecainide in Ventricular Arrhythmias" Encyclopedia, https://encyclopedia.pub/entry/14050 (accessed February 07, 2026).
Vetta, G. (2021, September 09). Flecainide in Ventricular Arrhythmias. In Encyclopedia. https://encyclopedia.pub/entry/14050
Vetta, Giampaolo. "Flecainide in Ventricular Arrhythmias." Encyclopedia. Web. 09 September, 2021.
Copy Citation
Flecainide is an IC antiarrhythmic drug (AAD) that received in 1984 Food and Drug Administration approval for the treatment of sustained ventricular tachycardia (VT) and subsequently for rhythm control of atrial fibrillation (AF). Currently, flecainide is mainly employed for sinus rhythm maintenance in AF and the treatment of idiopathic ventricular arrhythmias (IVA) in absence of ischaemic and structural heart disease on the basis of CAST data. Recent studies enrolling patients with different structural heart diseases demonstrated good effectiveness and safety profile of flecainide.
flecainide
flecainide controlled-release
class IC antiarrhythmic drug
CAST
ventricular arrhythmias
premature ventricular contraction
ventricular tachycardia
1. Introduction
Flecainide is a class IC antiarrhythmic drug (AAD) that was first synthesized in 1972. It was approved by Food and Drug Administration in 1984 for the treatment of symptomatic sustained Ventricular Tachycardia (VT), with a 90% efficacy and without significant adverse events [1][2]. Nowadays flecainide is mainly used for pharmacological conversion and sinus rhythm maintenance in patients with atrial fibrillation (AF) or supraventricular tachycardias [3][4]. The Cardiac Arrhythmia Suppression Trial (CAST) investigated the impact of class IC antiarrhythmic treatment on morbidity and mortality in patients with reduced ejection fraction and frequent premature ventricular complexes (PVC) after myocardial infarction (MI). Published in its preliminary form in 1989, CAST study recorded significantly higher mortality among patients treated with IC AADs compared to placebo and was prematurely dismissed [5].
The CAST study provided a major revision of the role of flecainide, which is therefore recommended in selected patients with preserved systolic function and without ischaemic heart disease [6]. Otherwise, flecainide is contraindicated in patients with previous MI even with preserved ejection fraction and should not be used in case of proved inducible ischaemia. In addition, the current guidelines extended the CAST findings to non-ischaemic structural heart disease despite limited evidence [6].
Thus, the CAST significantly affected and restricted the current use of flecainide in clinical practice causing a fall in employment of class IC AADs in favour of class III AADs [7][8] and exposing patients to the numerous adverse reactions and toxicities of these drugs, in the absence of robust evidence [9][10].
It is important to underline the paucity of available regarding patients with subcritical coronary artery disease (CAD) without previous MI or inducible ischemia with preserved systolic function and in patients with non-ischaemic structural heart disease. On the other side, recent studies demonstrated the safety and efficacy of flecainide in patients with extrasystole-induced cardiomyopathy or Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) and no Left Ventricular (LV) dysfunction [11][12][13][14].
2. Epidemiology of Ventricular Arrhythmia
VT is commonly seen in medical practice, not rarely in its most feared consequence, Sudden Cardiac Death (SCD). The American Heart Association (AHA) reports over 550,000 annual cardiac arrests, representing half of all cardiovascular deaths [15].
Most patients with sustained VT have underlying structural heart disease such as prior MI, dilated non-ischaemic cardiomyopathy, cardiac sarcoidosis and ARVC [15]. The overall incidence of sustained VT during an acute MI is approximately 10.2% with a high in-hospital mortality rate up to 27% [16]. VT in patients with structural heart disease often causes deleterious haemodynamic effects and is correlated with an increased risk of SCD, particularly in the elderly [17].
VT can also arise in otherwise normal hearts, Idiopathic Ventricular Arrhythmias (IVA); idiopathic PVCs are the most common arrhythmia in patients with normal heart: approximately 40% of adults experience PVCs on 24-h Holter monitoring [18]. IVAs may be classified into several subtypes according to the origin site. The ventricular outflow tracts are the most common origins of IVAs. Approximately 50% of IVAs originate from the Right Ventricular Outflow Tract (RVOT) [19]. Other origins include the Left Ventricular Outflow Tract (LVOT) (15%), the epicardial myocardium, the great cardiac veins and rarely the pulmonary artery [6][19].
3. Mechanisms and Clinical Evaluation of Ventricular Arrhythmia
3.1. Pathogenesis
Overall, three mechanisms could be the cause of ventricular arrhythmias: abnormal automaticity, triggered activity and reentry [22][23].
Abnormal automaticity refers to spontaneous depolarization of cells without pacemaker function, as working atrial and ventricular myocardial cells, due to a decrease of membrane potential. Abnormal automaticity is the leading cause of accelerated idioventricular rhythm and it’s caused by ischaemia, electrolyte disturbances and drugs increasing adrenergic tone [24].
The triggered activity results from early or delayed afterdepolarization in ventricular myocytes during phases 2 and 3 or phase 4 of the action potential. It occurs as a result of cyclic adenosine monophosphate dependent diastolic release of intracellular calcium; idiopathic RVOT tachycardia is an example of triggered activity [25][26].
Reentry is the most frequent mechanism causing serious ventricular arrhythmias and occurs when a PVC encounters a heterogeneously conductive substrate within the ventricular myocardium, consisting of two conduction pathways with a different conduction velocity and refractory period. Reentry is the postulated mechanism for VT occurring in a patient with an established scar from prior infarction [27]. VT in non-ischaemic cardiomyopathy is usually due to reentry and is mediated by idiopathic patchy intramural scar. In the case of hypertrophic cardiomyopathy, VT often originates from reentry circuits at the septal level or in the area of localization of hypertrophy at the level of the LV [28].
3.2. Clinical Presentation and Electrocardiographic Morphology
During IVAs haemodynamic impairment or serious symptoms are unusual, but patients can have palpitations, atypical chest pain or LV dysfunction. IVAs are distinguished by whether or not they originate from the outflow tracts and display specific electrocardiographic features [29].
The RVOT is the most common site of origin of idiopathic outflow tract ventricular arrhythmias and is characterized on electrocardiogram (ECG) by a positive QRS complex in the inferior leads and a Left Bundle Branch Block (LBBB) morphology. The origins of RVOT and LVOT can be distinguished on the ECG by the precordial R-wave transition from a predominate S wave to a predominate R wave (rS to Rs). Transitions before V3 indicate an LVOT source and after V3 indicate an RVOT source [29][30][31][32], as seen in Figure 1. When LBBB QRS morphology is observed and the precordial transition occurs at V3, comparison to the patient’s sinus rhythm precordial transition can be useful: a V2 transition ratio ≥ 0.60 suggests LVOT origin with high sensitivity and specificity [33][34].
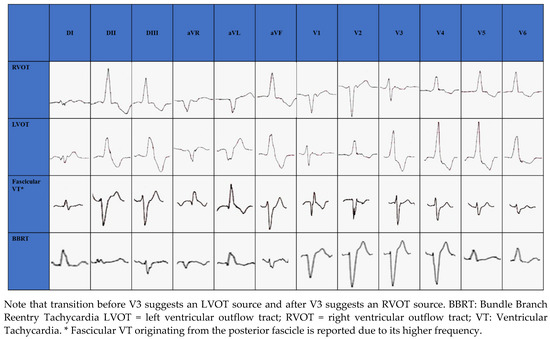
Figure 1. Electrocardiographic morphology of RVOT, LVOT, Fascicular VT and BBRT.
Among non-outflow tract IVAs, LV papillary muscles are a common site of origin. The postero-medial papillary muscle origin, which is the most common, is characterized by RBBB morphology with superior axis, while the antero-lateral papillary muscle origin is characterized by RBBB and inferior axis [35].
In structurally normal hearts, fascicular VT is generally caused by reentry circuit [36]; ECG morphology changes according to the exit site of the circuit from the retrograde-conducting fascicle: RBBB appearance with a left axis deviation if fascicular VT exits from the left posterior fascicle (90%) (Figure 1); RBBB appearance with a right axis deviation if VT exits from the left anterior fascicle [36]. Simultaneous anterograde activation of the left anterior and posterior fascicles with slow retrograde conduction through a separate septal fascicle can lead to upper septal fascicular VT (<1%), with narrow QRS (QRS < 110 ms) and normal axis [36][37].
BBRT is a macro reentrant tachycardia using the bundle branches as reentrant circuit pathways and is typically described in patients with bundle branch block. The most common morphology is the LBBB with a QRS ≥ 160 ms [6] (Figure 1).
4. Flecainide Pharmacology
4.1. Pharmacodynamics
Flecainide binds to open-state of fast inward Na+ channel Nav 1.5 in a rate and voltage-dependent manner [38]. The main result of flecainide is a reduction in the slope of phase 0 of monophasic action potential of His-Purkinje tissue and ventricular myocardium resulting in a slow-down of the conduction velocity with a prolongation of HV and QRS intervals, as seen in Figure 2. During a resting heart rate and in a healthy myocardium, the prolongation of HV and QRS is about 10%. The flecainide effect on slowing conduction velocity is proportional to heart rate [38]. Moreover, it is also very effective in areas already characterized by very slow conduction velocity. Usually, VTs with a low heart rate are caused by very slow conduction isthmuses and flecainide is very effective in these cases as it slows down conduction until the tachycardia is interrupted.
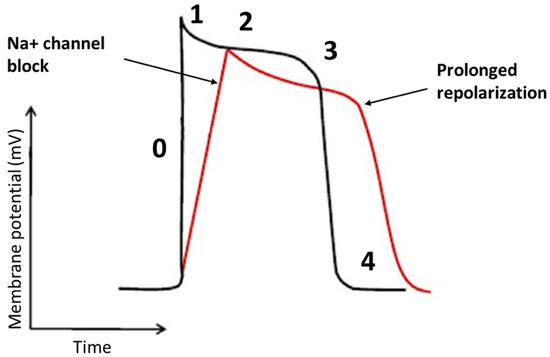
Figure 2. The effects of the flecainide on the cardiac action potential. The main effect of flecainide is a reduction in the slope of phase 0 with a minimum elongation of the action potential.
In addition, flecainide has an inhibitory action on the rapid component of the delayed rectifier K+ current and Kito channels [39]. Globally, flecainide increases the duration of the action potential and the effective refractory period in the myocardium, despite they are both reduced in the Purkinje system, due to the blockade of Na+ channels [40]. On the other hand, flecainide is highly efficient in the treatment of Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT), which is usually characterized by a high heart rate [41]. In this arrhythmia, however, the efficacy seems to be due to a different mechanism of action. Indeed, CPVT is induced by calcium overload due to sympathetic activation and to a diastolic calcium release from the sarcoplasmic reticulum through defective leaking ryanodine receptor (RYR) 2 channels. Flecainide blocks the RYR2 channels allowing direct targeting of the molecular defect [42].
The above mechanisms of action contribute to the efficacy of the drug in the treatment of ventricular arrhythmias, as seen in Figure 3.
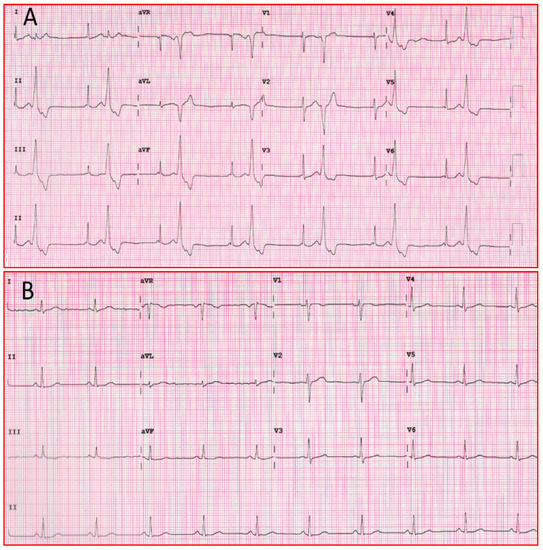
Figure 3. (A) Twelve-lead ECG of patient with ventricular bigeminism. (B) Twelve-lead ECG of the same patient one hour after taking a flecainide tablet. ECG: electrocardiogram.
Flecainide is also effective in Long QT Syndrome (LQT) type 3 as it inhibits not only the peak but also the late component of the Na+ current, causing a shortening of the QT interval. This form, indeed, is caused by mutations that increase the late Na+ current [43]. In addition, flecainide reduces Na+ and Ca2+ influx into myocardiocytes causing a negative inotropic effect and a reduction of cardiac output, particularly in patients with CAD or LV dysfunction [38].
Flecainide, due to its pharmacodynamics, widens the PR (17–29%), QT interval (3–8%) and QRS complex (11–27%) [44][45]. In 3–5% of cases flecainide causes conversion of AF to atrial flutter with a slow atrial rate leading to 1:1 atrioventricular conduction with high ventricular response [46]. However, this phenomenon can be prevented by concomitant therapy with negative dromotropic agents (beta-blockers, verapamil, diltiazem or digoxin) [47]. The main non-cardiac side effects are dizziness and visual disturbances as a result of the action of flecainide on Na channels, while headache and gastrointestinal disturbances are less frequent [48].
4.2. Pharmacokinetics
The gastrointestinal tract almost entirely absorbs flecainide, which has a bioavailability of 85–90% and achieves peak concentration in approximately 1–3 h [49]. Flecainide has a large volume of distribution with 40% of the drug bound to plasma proteins [49]. Therapeutic plasma levels range between 0.2 and 1.0 mg/mL, higher concentrations are associated with proarrhythmic side effects. Flecainide is metabolized by liver cytochromes CYP2D6 and CYP1A2 and then eliminated in urine [50]. The half-life is in the range of 12–27 h and may last up to 70 h in patients with heart failure, renal disease (creatinine clearance < 50 mL/min) liver disease and advanced age [44]. CYP2D6 genotype is a determinant factor of age-related decline in metabolic clearance of flecainide, resulting in a more prominent effect of the CYP2D6 genotype in the elderly [51]. Moreover, the therapeutic range of serum concentration is lower in SCN5A promoter haplotype B carriers than in the wild-type haplotype A homozygotes [52]. Concerning drug interactions, CYP2D6 inducers (carbamazepine, phenytoin, phenobarbital, primidone) increase the elimination rate of flecainide [53][54].On the other hand, CYP2D6 inhibitors as amiodarone, protease inhibitors (amprenavir, darunavir, fosamprenavir, indinavir, lopinavir, ritonavir, saquinavir, tipranavir) [55][56], selective serotonin reuptake inhibitors (citalopram, fluoxetine, paroxetine, sertraline) [57] and serotonin-norepinephrine reuptake inhibitors (duloxetine, venlafaxine) increase flecainide plasma concentration and half-life [58].
4.3. Controlled Release Flecainide
Controlled-release flecainide has a reduced and delayed maximum concentration (26 h) and fewer fluctuations in blood levels than the immediate-release form, allowing once-daily administration. The steady-state blood level is reached after 4–5 days and ranges from 0.27 to 0.33 mcg/mL, far from the blood level at risk of adverse reactions. Thus, the controlled-release form increases treatment compliance and lowers the risk of adverse events and interactions with other drugs while maintaining clinical efficacy [59].
References
- Hudak, J.M.; Banitt, E.H.; Schmid, J.R. Discovery and Development of Flecainide. Am. J. Cardiol. 1984, 53, 17–20.
- Hodges, M.; Haugland, J.M.; Granrud, G.; Conard, G.J.; Asinger, R.W.; Mikell, F.L.; Krejci, J. Suppression of Ventricular Ectopic Depolarizations by Flecainide Acetate, a New Antiarrhythmic Agent. Circulation 1982, 65, 879–885.
- Nieuwlaat, R.; Capucci, A.; Camm, A.J.; Olsson, S.B.; Andresen, D.; Davies, D.W.; Cobbe, S.; Breithardt, G.; Le Heuzey, J.-Y.; Prins, M.H.; et al. Atrial Fibrillation Management: A Prospective Survey in ESC Member Countries: The Euro Heart Survey on Atrial Fibrillation. Eur. Heart J. 2005, 26, 2422–2434.
- Lavalle, C.; Magnocavallo, M.; Straito, M.; Santini, L.; Forleo, G.B.; Grimaldi, M.; Badagliacca, R.; Lanata, L.; Ricci, R.P. Flecainide How and When: A Practical Guide in Supraventricular Arrhythmias. J. Clin. Med. 2021, 10, 1456.
- Echt, D.S.; Liebson, P.R.; Mitchell, L.B.; Peters, R.W.; Obias-Manno, D.; Barker, A.H.; Arensberg, D.; Baker, A.; Friedman, L.; Greene, H.L. Mortality and Morbidity in Patients Receiving Encainide, Flecainide, or Placebo. The Cardiac Arrhythmia Suppression Trial. N. Engl. J. Med. 1991, 324, 781–788.
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC)Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867.
- Anderson, J.L.; Pratt, C.M.; Waldo, A.L.; Karagounis, L.A. Impact of the Food and Drug Administration Approval of Flecainide and Encainide on Coronary Artery Disease Mortality: Putting “Deadly Medicine” to the Test. Am. J. Cardiol. 1997, 79, 43–47.
- Al-Khatib, S.M.; LaPointe, N.M.A.; Curtis, L.H.; Kramer, J.M.; Swann, J.; Honig, P.; Califf, R.M. Outpatient Prescribing of Antiarrhythmic Drugs from 1995 to 2000. Am. J. Cardiol. 2003, 91, 91–94.
- Ruzieh, M.; Moroi, M.K.; Aboujamous, N.M.; Ghahramani, M.; Naccarelli, G.V.; Mandrola, J.; Foy, A.J. Meta-Analysis Comparing the Relative Risk of Adverse Events for Amiodarone Versus Placebo. Am. J. Cardiol. 2019, 124, 1889–1893.
- Di Biase, L.; Romero, J.; Du, X.; Mohanty, S.; Trivedi, C.; Della Rocca, D.G.; Patel, K.; Sanchez, J.; Yang, R.; Alviz, I.; et al. Catheter Ablation of Ventricular Tachycardia in Ischemic Cardiomyopathy: Impact of Concomitant Amiodarone Therapy on Short- and Long-Term Clinical Outcomes. Heart Rhythm 2021, 18, 885–893.
- Hyman, M.C.; Mustin, D.; Supple, G.; Schaller, R.D.; Santangeli, P.; Arkles, J.; Lin, D.; Muser, D.; Dixit, S.; Nazarian, S.; et al. Class IC Antiarrhythmic Drugs for Suspected Premature Ventricular Contraction–Induced Cardiomyopathy. Heart Rhythm 2018, 15, 159–163.
- Ermakov, S.; Gerstenfeld, E.P.; Svetlichnaya, Y.; Scheinman, M.M. Use of Flecainide in Combination Antiarrhythmic Therapy in Patients with Arrhythmogenic Right Ventricular Cardiomyopathy. Heart Rhythm 2017, 14, 564–569.
- Ermakov, S.; Hoffmayer, K.S.; Gerstenfeld, E.P.; Scheinman, M.M. Combination Drug Therapy for Patients with Intractable Ventricular Tachycardia Associated with Right Ventricular Cardiomyopathy. Pacing Clin. Electrophysiol. 2014, 37, 90–94.
- Della Rocca, D.G.; Santini, L.; Forleo, G.B.; Sanniti, A.; Del Prete, A.; Lavalle, C.; Di Biase, L.; Natale, A.; Romeo, F. Novel Perspectives on Arrhythmia-Induced Cardiomyopathy: Pathophysiology, Clinical Manifestations and an Update on Invasive Management Strategies. Cardiol. Rev. 2015, 23, 135–141.
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Field, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS Guideline for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Circulation 2018, 138, e272–e391.
- Lo, R.; Chia, K.K.M.; Hsia, H.H. Ventricular Tachycardia in Ischemic Heart Disease. Card. Electrophysiol. Clin. 2017, 9, 25–46.
- Markman, T.M.; Nazarian, S. Treatment of Ventricular Arrhythmias: What’s New? Trends Cardiovasc. Med. 2019, 29, 249–261.
- Hingorani, P.; Karnad, D.R.; Rohekar, P.; Kerkar, V.; Lokhandwala, Y.Y.; Kothari, S. Arrhythmias Seen in Baseline 24-Hour Holter ECG Recordings in Healthy Normal Volunteers During Phase 1 Clinical Trials. J. Clin. Pharm. 2016, 56, 885–893.
- Latchamsetty, R.; Yokokawa, M.; Morady, F.; Kim, H.M.; Mathew, S.; Tilz, R.; Kuck, K.-H.; Nagashima, K.; Tedrow, U.; Stevenson, W.G.; et al. Multicenter Outcomes for Catheter Ablation of Idiopathic Premature Ventricular Complexes. JACC Clin. Electrophysiol. 2015, 1, 116–123.
- Caceres, J.; Jazayeri, M.; McKinnie, J.; Avitall, B.; Denker, S.T.; Tchou, P.; Akhtar, M. Sustained Bundle Branch Reentry as a Mechanism of Clinical Tachycardia. Circulation 1989, 79, 256–270.
- Bhatt, A.G.; Mittal, S. Ventricular Tachycardia in Structurally Normal Hearts. In Encyclopedia of Cardiovascular Research and Medicine; Elsevier: Cham, Switzerland, 2018; pp. 700–724. ISBN 978-0-12-805154-2.
- Wit, A.L. Cellular Electrophysiologic Mechanisms of Cardiac Arrhythmias. Cardiol. Clin. 1990, 8, 393–409.
- Cabo, C.; Wit, A.L. Cellular Electrophysiologic Mechanisms of Cardiac Arrhythmias. Cardiol. Clin. 1997, 15, 517–538.
- Flinders, D.C.; Roberts, S.D. Ventricular Arrhythmias. Prim. Care 2000, 27, 709–724;vii.
- AlMahameed, S.T.; Ziv, O. Ventricular Arrhythmias. Med. Clin. N. Am. 2019, 103, 881–895.
- Lerman, B.B.; Ip, J.E.; Shah, B.K.; Thomas, G.; Liu, C.F.; Ciaccio, E.J.; Wit, A.L.; Cheung, J.W.; Markowitz, S.M. Mechanism-Specific Effects of Adenosine on Ventricular Tachycardia. J. Cardiovasc. Electrophysiol. 2014, 25, 1350–1358.
- Tschabrunn, C.M.; Roujol, S.; Nezafat, R.; Faulkner-Jones, B.; Buxton, A.E.; Josephson, M.E.; Anter, E. A Swine Model of Infarct-Related Reentrant Ventricular Tachycardia: Electroanatomic, Magnetic Resonance, and Histopathological Characterization. Heart Rhythm 2016, 13, 262–273.
- Bhaskaran, A.; De Silva, K.; Rao, K.; Campbell, T.; Trivic, I.; Bennett, R.G.; Kizana, E.; Kumar, S. Ventricular Tachycardia Ablation in Non-Ischemic Cardiomyopathy. Korean Circ. J. 2020, 50, 203–219.
- Lavalle, C.; Mariani, M.V.; Piro, A.; Straito, M.; Severino, P.; Della Rocca, D.G.; Forleo, G.B.; Romero, J.; Di Biase, L.; Fedele, F. Electrocardiographic Features, Mapping and Ablation of Idiopathic Outflow Tract Ventricular Arrhythmias. J. Interv. Card. Electrophysiol. 2020, 57, 207–218.
- Park, K.-M.; Kim, Y.-H.; Marchlinski, F.E. Using the Surface Electrocardiogram to Localize the Origin of Idiopathic Ventricular Tachycardia. Pacing Clin. Electrophysiol. 2012, 35, 1516–1527.
- Della Rocca, D.G.; Gianni, C.; Mohanty, S.; Trivedi, C.; Di Biase, L.; Natale, A. Localization of Ventricular Arrhythmias for Catheter Ablation: The Role of Surface Electrocardiogram. Card. Electrophysiol. Clin. 2018, 10, 333–354.
- Di Biase, L.; Romero, J.; Zado, E.S.; Diaz, J.C.; Gianni, C.; Hranitzki, P.M.; Sanchez, J.E.; Mohanty, S.; Al-Ahmad, A.; Mohanty, P.; et al. Variant of Ventricular Outflow Tract Ventricular Arrhythmias Requiring Ablation from Multiple Sites: Intramural Origin. Heart Rhythm 2019, 16, 724–732.
- Betensky, B.P.; Park, R.E.; Marchlinski, F.E.; Hutchinson, M.D.; Garcia, F.C.; Dixit, S.; Callans, D.J.; Cooper, J.M.; Bala, R.; Lin, D.; et al. The V(2) Transition Ratio: A New Electrocardiographic Criterion for Distinguishing Left from Right Ventricular Outflow Tract Tachycardia Origin. J. Am. Coll. Cardiol. 2011, 57, 2255–2262.
- Chen, Q.; Xu, J.; Gianni, C.; Trivedi, C.; Della Rocca, D.G.; Bassiouny, M.; Canpolat, U.; Tapia, A.C.; Burkhardt, J.D.; Sanchez, J.E.; et al. Simple Electrocardiographic Criteria for Rapid Identification of Wide QRS Complex Tachycardia: The New Limb Lead Algorithm. Heart Rhythm 2020, 17, 431–438.
- Dukkipati, S.R.; Koruth, J.S.; Choudry, S.; Miller, M.A.; Whang, W.; Reddy, V.Y. Catheter Ablation of Ventricular Tachycardia in Structural Heart Disease: Indications, Strategies, and Outcomes-Part II. J. Am. Coll. Cardiol. 2017, 70, 2924–2941.
- Sung, R.K.; Boyden, P.A.; Scheinman, M. Cellular Physiology and Clinical Manifestations of Fascicular Arrhythmias in Normal Hearts. JACC Clin. Electrophysiol. 2017, 3, 1343–1355.
- Sung, R.K.; Kim, A.M.; Tseng, Z.H.; Han, F.; Inada, K.; Tedrow, U.B.; Viswanathan, M.N.; Badhwar, N.; Varosy, P.D.; Tanel, R.; et al. Diagnosis and Ablation of Multiform Fascicular Tachycardia. J. Cardiovasc. Electrophysiol. 2013, 24, 297–304.
- Josephson, M.A.; Ikeda, N.; Singh, B.N. Effects of Flecainide on Ventricular Function: Clinical and Experimental Correlations. Am. J. Cardiol. 1984, 53, 95–100.
- Anno, T.; Hondeghem, L.M. Interactions of Flecainide with Guinea Pig Cardiac Sodium Channels. Importance of Activation Unblocking to the Voltage Dependence of Recovery. Circ. Res. 1990, 66, 789–803.
- Follmer, C.H.; Colatsky, T.J. Block of Delayed Rectifier Potassium Current, IK, by Flecainide and E-4031 in Cat Ventricular Myocytes. Circulation 1990, 82, 289–293.
- Padfield, G.J.; AlAhmari, L.; Lieve, K.V.V.; AlAhmari, T.; Roston, T.M.; Wilde, A.A.; Krahn, A.D.; Sanatani, S. Flecainide Monotherapy Is an Option for Selected Patients with Catecholaminergic Polymorphic Ventricular Tachycardia Intolerant of β-Blockade. Heart Rhythm 2016, 13, 609–613.
- Hilliard, F.A.; Steele, D.S.; Laver, D.; Yang, Z.; Le Marchand, S.J.; Chopra, N.; Piston, D.W.; Huke, S.; Knollmann, B.C. Flecainide Inhibits Arrhythmogenic Ca2+ Waves by Open State Block of Ryanodine Receptor Ca2+ Release Channels and Reduction of Ca2+ Spark Mass. J. Mol. Cell. Cardiol. 2010, 48, 293–301.
- Belardinelli, L.; Giles, W.R.; Rajamani, S.; Karagueuzian, H.S.; Shryock, J.C. Cardiac Late Na+ Current: Proarrhythmic Effects, Roles in Long QT Syndromes, and Pathological Relationship to CaMKII and Oxidative Stress. Heart Rhythm 2015, 12, 440–448.
- Holmes, B.; Heel, R.C. A Preliminary Review of Its Pharmacodynamic Properties and Therapeutic efficiancy. Drugs 1985, 29, 1–33.
- Roden, D.M.; Woosley, R.L. Drug Therapy. Flecainide. N. Engl. J. Med. 1986, 315, 36–41.
- Crijns, H.J.; van Gelder, I.C.; Lie, K.I. Supraventricular Tachycardia Mimicking Ventricular Tachycardia during Flecainide Treatment. Am. J. Cardiol. 1988, 62, 1303–1306.
- Boriani, G.; Diemberger, I.; Biffi, M.; Martignani, C.; Branzi, A. Pharmacological Cardioversion of Atrial Fibrillation: Current Management and Treatment Options. Drugs 2004, 64, 2741–2762.
- Gentzkow, G.D.; Sullivan, J.Y. Extracardiac Adverse Effects of Flecainide. Am. J. Cardiol. 1984, 53, 101–105.
- Conard, G.J.; Ober, R.E. Metabolism of Flecainide. Am. J. Cardiol. 1984, 53, 41–51.
- Tjandra-Maga, T.; Verbesselt, R.; Hecken, A.; Mullie, A.; Schepper, P. Flecainide: Single and Multiple Oral Dose Kinetics, Absolute Bioavailability and Effect of Food and Antacid in Man. Br. J. Clin. Pharmacol. 1986, 22, 309–316.
- Zhou, S.-F. Polymorphism of Human Cytochrome P450 2D6 and Its Clinical Significance: Part I. Clin. Pharmacokinet. 2009, 48, 689–723.
- Doki, K.; Homma, M.; Kuga, K.; Aonuma, K.; Kohda, Y. SCN5A Promoter Haplotype Affects the Therapeutic Range for Serum Flecainide Concentration in Asian Patients. Pharm. Genom. 2013, 23, 349–354.
- Aliot, E.; Capucci, A.; Crijns, H.J.; Goette, A.; Tamargo, J. Twenty-Five Years in the Making: Flecainide Is Safe and Effective for the Management of Atrial Fibrillation. Europace 2011, 13, 161–173.
- Tamargo, J.; Capucci, A.; Mabo, P. Safety of Flecainide. Drug Saf. 2012, 35, 273–289.
- Shea, P.; Lal, R.; Kim, S.S.; Schechtman, K.; Ruffy, R. Flecainide and Amiodarone Interaction. J. Am. Coll. Cardiol. 1986, 7, 1127–1130.
- Meda Pharmaceuticals Ltd. Flecainide Acetate—Summary of Product Characteristics (UK). 2013. Available online: www.Medicines.Org.Uk/Emc/Medicine/3905 (accessed on 17 August 2014).
- Lim, K.S.; Cho, J.-Y.; Jang, I.-J.; Kim, B.-H.; Kim, J.; Jeon, J.-Y.; Tae, Y.-M.; Yi, S.; Eum, S.; Shin, S.-G.; et al. Pharmacokinetic Interaction of Flecainide and Paroxetine in Relation to the CYP2D6*10 Allele in Healthy Korean Subjects. Br. J. Clin. Pharm. 2008, 66, 660–666.
- Nemeroff, C.B.; DeVane, C.L.; Pollock, B.G. Newer Antidepressants and the Cytochrome P450 System. Am. J. Psychiatry 1996, 153, 311–320.
- Coumel, P.; Maison-Blanche, P.; Tarral, E.; Périer, A.; Milliez, P.; Leenhardt, A. Pharmacodynamic Equivalence of Two Flecainide Acetate Formulations in Patients with Paroxysmal Atrial Fibrillation by QRS Analysis of Ambulatory Electrocardiogram. J. Cardiovasc. Pharmacol. 2003, 41, 771–779.
More
Information
Subjects:
Pharmacology & Pharmacy
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.5K
Revisions:
2 times
(View History)
Update Date:
10 Sep 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No