Transcription factors represent a large class of proteins with more than 1500 members, of which 15%–20% are considered oncogenes
[1][2]. They are classified into more than 70 different families based on sequence and structure homologies of their DNA-binding domains. Among them, the homeobox DNA binding domain (homeodomain, HD) family encompasses more than 250 transcription factors, themselves subdivided into 21 different sub-families, including HOX-Like, Para-HOX, NK-Like, TALE (three amino acid loop extension) containing the PBC and MEIS groups among other), POU, HNF, LIM, Paired (PRD) and PRD-like subfamilies
[3][4]. Derived from
Antennapedia and
Bithorax ancestors, the HOX-Like subgroup corresponds to the HOX cluster genes and is the only group of HD proteins conventionally named “HOX genes.” Organized into four paralog clusters in animals, the number and identity of HOX genes varies depending on the species. In humans, 39 HOX proteins are organized from 1 to 13 (as originally defined in
Drosophila) in the four paralog clusters A to D. After their spatiotemporal expression, critical for patterning of the anterior–posterior axis in embryos, HOX gene expression is largely repressed in adults but controlled reactivation allows dynamic expression in adults to lead or reactivate some cellular processes, including hematopoiesis
[5][6][7][8], wound healing
[9][10], vascularization, endometrial development/fertility
[11][12][13][14][15] and many other processes of body repair and homeostasis
[16][17][18][19][20]. In particular, HOXA9 expression is finely controlled and decreases during the progression of normal hematopoiesis
[21].
A large proportion of HOX cluster genes are considered oncogenes due to their implication in translocations, mutations or improper expression dynamics in some tissues. They are associated with cancer at the initiation, development or metastasis stages and are over-expressed in cancer cells relative to normal tissue (for reviews
[22][23][24][25][26][27][28][29]). As an example, the oncogenic function of HOXA cluster genes was originally well described in hematopoietic disorders. This is particularly the case of HOXA9 in acute myeloid leukemia (AML).
2. HOXA9: A Leukemic Driver in AML
The leukemogenic function of HOXA9 was first assessed in the murine model BXH-2, a mouse strain that spontaneously develops AML through endogenous retroviral integration. Indeed, Hoxa9, as well as a number of other Hox genes and its co-factor Meis1 (myeloid ecotropic viral integration site 1), are frequently over-expressed in BXH-2 murine leukemic cells
[30]. It was then evidenced that transplantation of cells over-expressing murine Hoxa9 by retroviral transduction evidenced a late onset of AML, a process that was accelerated by co-transduction with Meis1
[31].
In human leukemia, the first implication of HOXA9 was highlighted by the discovery of the NUP98-HOXA9 fusion protein resulting from t(7;11)(p15;p15) translocation
[30][32], a rare (1%–3%) AML subtype associated with poor prognosis
[33][34]. NUP98, a nucleoporin of 98 kDa, is a chaperone protein associated with the nuclear pore. NUP98-HOXA9 binds directly to DNA on the HOXA9-cognate sequence via HOXA9 homeodomain
[35]. NUP98-HOXA9 chimera seems to induce myelodysplastic syndromes for a relatively long period before transformation in AML, a period that is reduced when MEIS1 is concomitantly expressed
[36]. The NUP98-HOXA9 protein would also have a higher transcriptional activity than HOXA9 itself due to its highest stability (half-life three times longer) in relation to its resistance to ubiquitinylation mediated by CUL-4A that may partly explain its oncogenic function
[37].
Besides expressed as an oncogenic fused protein, the implication of structurally unmodified HOXA9 as a leukemic driver was then evidenced as part of its over-expression in leukemic cells. Indeed, the relevance of HOXA9 expression in global survival of human leukemia patients was first demonstrated on gene expression signatures relating to patient outcome: HOXA9 was evidenced as the protein presenting the highest correlation with poor prognosis in a series of nearly 7000 genes
[38]. This association of the level of HOXA9 expression with prognosis was also evaluated on an independent series of patients showing that low levels of HOXA9 (but also other HOXA and HOXB) gene expression is characteristic of a favorable cytogenetic AML subgroup
[39][40]. Such general analyses now take into account the better knowledge of cytogenetic and molecular characteristics of different AML sub-types and it is now well established that HOXA9 over-expression is directly associated with some of them, themselves directly identified as good, intermediate, or adverse prognosis subgroups, with a total prevalence of ~70% of AML
[41][42]. The main genetic alterations associated with HOXA9 over-expression in AML are presented in
Table 1.
Table 1. List of the main genetic alterations in AML that are associated with HOXA9 over-expression.
| Type of Alteration |
Fusion/Mutation/Additional Chromosome |
Translocation/Inversion/Deletion |
References |
| Chromosomal alterations |
MLL fusions |
11q23 translocations |
[43][44][45] |
| NUP98-NSD1 |
t(5;11)(q35;p15) |
[46][47] |
| NUP98-HOXA9 |
t(7;11)(p15;p15) |
[48] |
| NUP98-HOXA10 |
t(7;11)(p15;p15) |
[49] |
| NUP98-HOXC11 |
t(11;12)(p15;q13) |
[50] |
| NUP98-HOXD11 |
t(2;11)(q31;p15) |
[51] |
| NUP98-HOXD13 |
t(2;11)(q31;p15) |
[49] |
| NUP98-HHEX |
t(10;11)(q23;p15) |
[52] |
| NUP98-KDM5A |
t(11;12)(p15;p13) |
[33] |
| NUP98-PHF23 |
t(11;17)(p15;p13) |
[33] |
| NUP98-PRRX1 |
t(1;11)(q24;p15) |
[33] |
| NUP98-DDX10 |
inv(11)(p15q22) |
[33] |
| MYST3-CREBBP |
t(8;16)(p11;p13) |
[53] |
| RUNX1-EVI1 |
t(3;21)(q26;q22) |
[54] |
| CDX2-ETV6 |
t(12;13)(p13;q12) |
[55] |
| CALM-AF10 |
t(10;11)(p12-14;q14-21) |
[56] |
| SET-NUP214 |
del(9)(q34.11;q34.13) |
[57] |
| NPM1-MLF1 |
t(3;5)(q25;q34) |
[58][59] |
| +8 |
/ |
[60] |
| Mutations |
NPM1 |
|
[61][62][63][64] |
| MLL-PTD |
[42] |
| DNMT3A |
[65] |
| EZH2 |
[42] |
| IDH1/2 |
[63][66] |
| Polymorphism |
GFI1-S36N |
|
[67] |
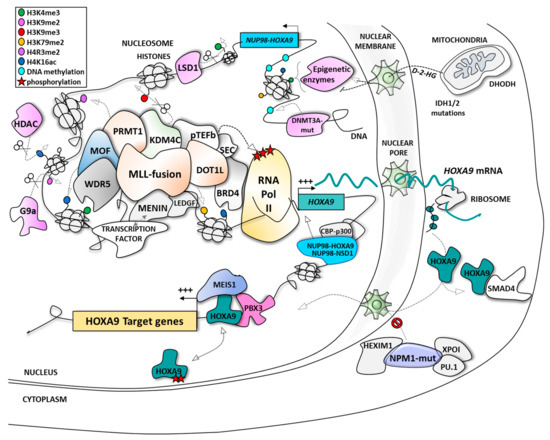
The most described HOXA9-associated leukemias are: (1) acute leukemia (either myeloid or lymphoid) bearing MLL (mixed lineage leukemia, also called KMT2A) fusions
[43][68][44][69][45], known as mixed phenotype acute leukemia (MPAL), and which represent ~5% of AML and are associated with poor prognosis; and (2) AML with nucleophosmin 1 (NPM1) mutations, which represent ~55% of normal karyotype AML and ~35% of all AMLs, and are associated with poor to intermediate prognosis depending on the nature of additional alterations, such as mutations of FLT3 kinase (Fms-like tyrosine kinase 3)
[61][62][63].
The AML subtype MPAL preferentially affects infants or is developed as a therapy-induced leukemia. MPAL is associated with poor prognosis with a five-year survival rate of less than 40% in infants compared to ~90% for non-MPAL
[70]. The genomic breakpoints involve more than 130 different MLL translocation partners already described, with the 10 main partners representing >90% of the MLL translocations, including AF9 (~30%), AF10 (~16%), ELL (~10%), AF6 (~8%), and ENL (~6%)
[71][72][73]. The major breakpoint cluster region is localized between exon 9 and intron 11 of the MLL gene in more than 80% of MPAL patients. These rearrangements generate a fusion between the N-terminal portion of the MLL protein containing its DNA binding domain and the carboxy-terminal portion of its protein partner
[74]. The MLL protein will lose its SET domain and its domain for binding to ASB2, a ubiquitin ligase causing its proteolysis. Thereby, the fusion proteins generated will no more be degraded
[75]. Interestingly, the main translocation partners (AF9/AF10/ENL), as well as minor partners such as AF4, are proteins that normally function within a large protein complex associated with the MLL protein (within a large complex or different sub-complexes). Translocations seem to physically fix proteins together in order to favor the stability and functionality of the MLL complex, particularly through interaction (direct or indirect) with the disruptor of telomeric silencing 1-like protein DOT1L (through direct interaction with AF10, for instance), an epigenetic partner that methylates lysine-79 residues of histone H3 proteins as a transcriptional activation mark
[76][77][78], or with p-TEFb kinase (through direct interaction with AF4, for instance) that phosphorylates RNA polymerase II to allow gene transcription
[79]. Among other proteins implicated in the active MLL complex are Menin
[80][81], LEDGF (lens epithelium-derived growth factor)
[80][82], WDR5 (WD repeat protein 5)
[83], BRD4 (bromodomain-related protein 4)
[84], HDAC (histone deacetylase)
[85][86], KDM4C/JMJD2C (lysine-specific demethylase 4C/jumonji domain-containing protein 2C) and PRMT1 (protein arginine N-methyltransferase 1)
[87] (
Figure 1).
Figure 1. The different modes of regulation of HOXA9 expression and function in acute myeloid leukemia (AML). BRD4, bromodomain-related protein 4; CBP, CREB-binding protein; CDK9, cyclin-dependent kinase 9; D-2-HG, D-2-hydroxyglutarate; DHODH, dihydroorotate dehydrogenase; DNMT3A, DNA methyl transferase 3A; DOT1L, disruptor of telomeric silencing 1-like protein; HDAC, histone deacetylase; HEXIM1, hexamethylene bisacetamide (HMBA) inducible protein 1; HOXA9, homeobox A9; IDH, isocitrate dehydrogenase; KDM4C/lysine-specific demethylase 4C; LEDGF, lens epithelium-derived growth factor; LSD1, lysine-specific demethylase 1; MEIS1, myeloid ecotropic viral integration site 1; MLL, mixed lineage leukemia; MOF, males absent on the first; NPM1, nucleophosmin 1; NSD1, nuclear receptor binding SET domain protein 1; NUP98, nucleoporin 98kDa; PBX3, pre-B-cell leukemia transcription factor 3; PRMT1, protein arginine N-methyltransferase 1; pTEFb, positive transcription elongation factor b; SMAD4, mothers against decapentaplegic homolog 4; WDR5, WD repeat protein 5; XPO-1, exportin-1.
In mice, grafting of bone marrow cells with retroviral transduction of MLL fusion proteins deregulated the expression of Hox genes
[88]. All MPAL patients not only evidenced HOXA9 over-expression but also middle HOXA cluster over-expression, as exemplified by MLL-AF9 fusion, which positively regulates the expression of HOXA6, HOXA7, HOXA9, and HOXA10
[89]. MLL translocations also increase the expression of MEIS1, which is generally positively correlated with HOXA9 expression
[29]. In addition, although the role of an alternative transcript HOXA9T is not clearly defined, HOXA9T is over-expressed in AMLs with MLL arrangements
[90]. Beside these fusion proteins, partial tandem duplications (MLL-PTD) were discovered, particularly in de novo AML cases. The most common duplication event is a copy of exons 5-11 or 5-12 inserted into intron 4 and resulting in the replication of the N-terminal portion of MLL that contains the AT-hook DNA binding domain. These alterations represent 12% of AML
[72] and are associated with poor prognosis. By contrast, MPAL with MLL-PTD evidences moderate up-regulation of HOXA9 expression
[91]; however, HOXA9 is still crucial for MLL-PTD-driven leukemogenic processes
[42][92].
The NPM1 mutations are generally present in de novo adult AML with normal karyotype and were evidenced to be correlated with the over-expression of HOXA, MEIS1, and FLT3 genes
[61][93]. Under normal conditions, NPM1 chaperone protein is located in the nucleus. NPM1 mutations result in the delocalization of the protein into the cytoplasm causing over-expression of HOXA9, HOXA10, and MEIS1
[62] by a mechanism probably associating MLL, P-TEFb, DOT1L, and/or menin
[93][64]. One of the suggested mechanisms for mutated-NPM1 (also called NPM1c+) control of HOXA9 expression is the activation of the transcriptional complex P-TEFb (positive transcription elongation factor b), a partner of the MLL complex usually sequestrated by HEXIM1 in the cytoplasm; HEXIM1 (hexamethylene bisacetamide (HMBA) inducible protein 1) sequestrated in the cytoplasm by mutated-NPM1 could no longer interact with P-TEFb, which could therefore activate the MLL complex and subsequently HOXA9 expression
[94][95] (
Figure 2). Recently, it was also highlighted that NPM1c+ leukemic cell survival requires upregulation of HOXA9 and its DNA-binding partner, the Pre-B-cell leukemia homeobox 3 PBX3 in a MLL/DOT1L dependent manner
[64].
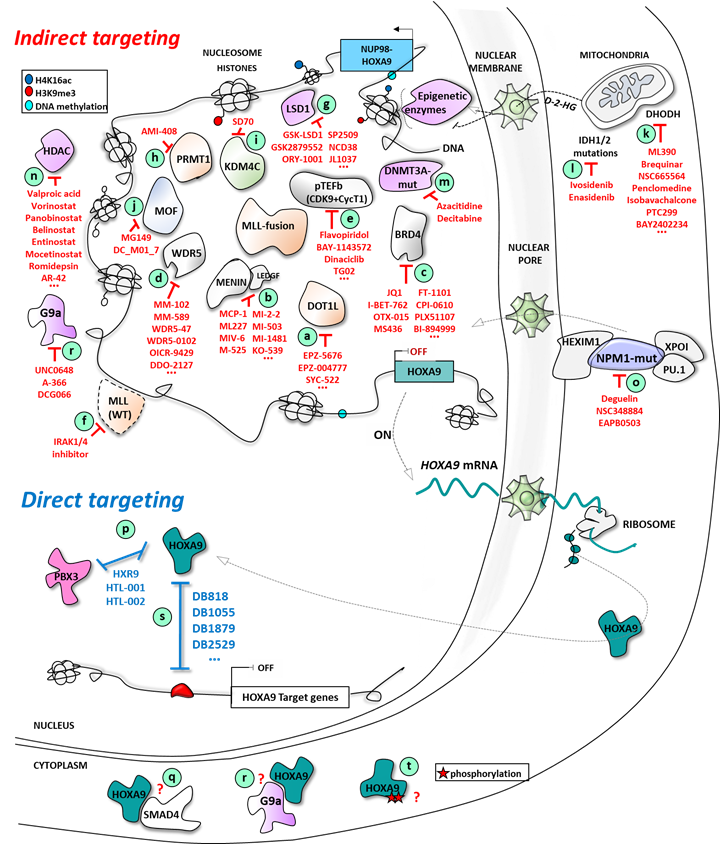
Figure 2. Direct and indirect targeting of HOXA9 expression in AML: multiple epigenetic and non-epigenetic therapeutic opportunities.
In parallel, Brunetti et al. demonstrated that HOXA (and HOXB) genes are not only direct downstream targets of NPM1c+ protein but also that the interaction between exportin-1 (XPO-1), a nuclear pore exporter, and NPM1c+ protein, maintains mutated-NPM1 in the cytoplasmic compartment as an important point explaining AML occurrence
[96]. Moreover, Gu et al.
[97] evidenced NPM1c+ interaction with PU.1/SPI1 transcription factor as another way to maintain NPM1 within this complex in the cytoplasm, whereas PU.1 over-expression is associated with a decrease in HOXA gene expression.
The over-expression of HOXA9 is also found in many NUP98 fusions containing AML samples, accounting for a total of 1%–3% of AML
[33]. Some examples are presented in
Table 1. A lot of these partners are associated with epigenetic control or the DNA binding function (with, for instance, a lot of homeodomain containing transcription factor partners). The most frequent of these is the H3K36 methyltransferase NSD1 (nuclear receptor binding SET domain protein 1), forming the NUP98-NSD1 fusion protein that activates HOXA gene expression for leukemogenesis in 1%–2% AML
[46].
Trisomy 8 (+8) represents ~10% of all AML and also correlates with a high level of HOXA9 expression
[60]. Alone, trisomy 8 is not sufficient for leukemogenesis but is often associated with the t(7;12) or t(9;11) and t(1;11) MLL translocations. HOXA9 and HOXA10 are the first and second rated over-expressed genes in +8 AML, respectively, relative to normal bone marrow cells
[60]. However, this analysis does not exclude MPAL and +8 double positive AML, and further analysis may be required to ensure that MLL alteration was not the main driver of HOXA9/10 over-expression in +8 AML.
In many other well-defined cytogenetic or molecular alterations associated with AML, HOXA9 can be frequently over-expressed but not in all patients of the same sub-group, suggesting that further analysis of additional alteration would be required. For instance, EVI1 over-expressing leukemia results from t(3;21)(q26;q22) associated with poor survival in 8%–10% AML and presents over-expression of the HOXA9 gene with a large spread of HOXA9 expression from positive to negative.
In total, the proportion of HOXA9-over-expressed AML is ~70%. This high proportion highlights HOXA9 as an interesting potential target to treat such AML.
The oncogenic function of HOXA9 in AML is associated with cell proliferation, differentiation blockade, increased malignancy of leukemic cells, and progenitor self-renewal maintenance
[98]. Invalidation of HOXA9 expression in those cells impairs proliferation and leukemic properties, and re-activates differentiation processes, showing that HOXA9 is a functional target to restore differentiation in AML
[45][99][100][101]. More precisely, the presence of the HOXA9 DNA binding domain is a prerequisite for HOXA9-induced leukemic transformation in mice models: (i) swapping HOXA9 homeodomain with HOXA1 homeodomain in the HOXA9 transcription factor is sufficient to abolish the leukemic potential of transduced murine hematopoietic progenitor cells engrafted in mice, whereas transferring HOXA9 homeodomain in the HOXA1 protein maintains the leukemic propensity of HOXA9 and results in common deregulated gene signatures with wild-type HOXA9-induced transformation
[102]; (ii) mutating HOXA9 homeodomain at Asn51 to a serine residue (N51S) abolishes leukemic transformation in mice
[103][104]. Similarly, HOXA9T, a splice variant protein which has lost its DNA binding domain, does not induce leukemia by itself, even if it seems to support the leukemogenic activity of HOXA9
[105] by a yet unclear mechanism of action, but may imply HOXA9T binding to transcription promoting factors such as CBP (CREB-binding protein) or some chaperone proteins
[106]. Interestingly, the phosphorylation status of HOXA9 changes its DNA binding activity and consequently its propensity to induce leukemia, as demonstrated with protein kinase C (PKC) phosphorylation of Ser204 of the HOXA9 DNA binding domain, impairing DNA binding and leading to myeloid differentiation of murine Hoxa9-immortalized bone marrow cells
[107].
In order to understand HOXA9-mediated leukemogenesis, the key point would be to identify the network underlying its transcriptional activity using ChIP-sequencing. However, due to the lack of ChIP and ChIP-seq grade antibodies directed against HOXA9 and other HOX proteins to identify endogenous targets for each HOX protein on the chromatin, most global ChIP analyses have used exogenous expression of tagged proteins. This is notably the case of a HA-tagged Hoxa9 protein in a Hoxa9- and Meis1-transformed murine bone marrow cell model to identify thousands of genomic binding regions of the murine Hoxa9 transcription factor, being associated or not with Meis. These studies identified several pro-leukemic Hoxa9 target genes such as Erg (ETS-related gene), Flt3, Lmo2 (LIM domain only 2), and c-Myb (myeloblastosis)
[108]. However, a recent study has shown that Hoxa9 is a specific substrate of a granule protease and that its inhibition would allow the ChIP-sequencing analysis in primary transformed murine cells, showing a feedback loop driving expression of key oncogenes and cell cycle control genes
[109].
Alternatively, microarray or RNA-seq analyzes have been successfully performed. Gene expression analyses in models over-expressing or interfering (shRNA, CRISPR9-Cas9) with HOXA9 expression showed significant transcriptomic modulations in which HOXA9 could act as an activator or a repressor, depending on target gene and cell context
[110]. Most HOXA9-specific targets were also discovered individually, including Lmo2, Bcl-2, Fgf2, Igf1, Ink4a/b, and c-Myb
[108][110][111][112][113][114][115][116]. In particular, HOXA9 functions as a pioneer factor at de novo enhancers and recruits CEBPα and the MLL3/MLL4 complex
[117]. HOXA9 over-expression in progenitor cells, therefore, leads to significant enhancer reorganizations with prominent emergence of leukemia-specific de novo enhancers.
If HOXA9 could act alone to trigger leukemia, it requires cofactors to increase its propensity to induce leukemia. For HOXA9, the leukemogenic activity of MEIS1 was discovered through in vivo experiments in BXH-2 mice, a pro-viral insertion model in which 15% of induced AMLs are caused by pro-viral insertion into the Meis1 gene locus
[118]. Like HOXA9, Meis1 expression decreases during normal differentiation of blood cells. In vivo, the presence of Hoxa9 expressed alone in murine bone marrow cells is not sufficient to rapidly induce leukemia (>6 months) whereas concomitant expression of Hoxa9 with Meis1 greatly shortened this period (approximately 67 days). However, Meis1 alone does not induce leukemia
[31], Meis1 as an accelerator of Hoxa9-induced leukemia but not stricto sensu as an oncogene. In human AML samples, a correlation expression of MEIS1 and HOXA9 is observed, suggesting a parallel or common temporal action of these factors
[30][119][120]. If MEIS1 potentiates the leukemia action of HOXA9, this is also the case of PBX3, whose expression is also strongly correlated with HOXA9 expression, especially in leukemia subtypes with a normal karyotype or associated with MLL rearrangements. The overexpression of PBX3 and HOXA9 thus favors the initiation and implantation of AML
[29]. Inactivation of PBX3 and HOXA9 by down-regulating H3K79 methylation also represses NPM1c+ leukemic cell survival
[64].
As HOXA9 is associated with a large proportion of AML, its inhibition is an interesting strategy against AML that could be achieved by different ways:
-
inhibition of its expression;
-
blockade of the specific protein/protein interaction crucial for its mechanism of action;
-
or, more specifically as part of a transcription factor, the blockade of the interaction with its cognate sequence on the DNA.
 +1 credit
+1 credit