HOXA9 (Homeobox A9) is a homeotic transcription factor known for more than two decades to be associated with leukemia. The expression of HOXA9 homeoprotein is associated with anterior–posterior patterning during embryonic development, and its expression is then abolished in most adult cells, with the exception of hematopoietic progenitor cells. The oncogenic function of HOXA9 was first assessed in human acute myeloid leukemia (AML), particularly in the mixed-phenotype associated lineage leukemia (MPAL) subtype. HOXA9 expression in AML is associated with aggressiveness and a poor prognosis. Since then, HOXA9 has been involved in other hematopoietic malignancies and an increasing number of solid tumors. Despite this, HOXA9 was for a long time not targeted to treat cancer, mainly since, as a transcription factor, it belongs to a class of protein long considered to be an “undruggable” target; however, things have now evolved.
- HOXA9
- acute myeloid leukemia
- transcription factor
- epigenetic
- protein/protein interaction inhibitors
- protein/DNA interaction inhibitors
1. Introduction
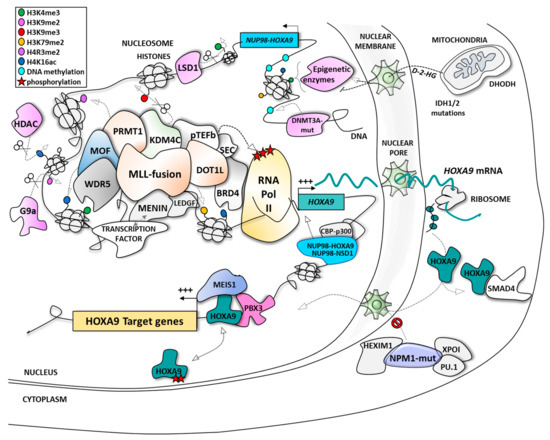
2. HOXA9: A Leukemic Driver in AML
| Type of Alteration | Fusion/Mutation/Additional Chromosome | Translocation/Inversion/Deletion | References |
|---|---|---|---|
| Chromosomal alterations | MLL fusions | 11q23 translocations | [43][44][45] |
| NUP98-NSD1 | t(5;11)(q35;p15) | [46][47] | |
| NUP98-HOXA9 | t(7;11)(p15;p15) | [48] | |
| NUP98-HOXA10 | t(7;11)(p15;p15) | [49] | |
| NUP98-HOXC11 | t(11;12)(p15;q13) | [50] | |
| NUP98-HOXD11 | t(2;11)(q31;p15) | [51] | |
| NUP98-HOXD13 | t(2;11)(q31;p15) | [49] | |
| NUP98-HHEX | t(10;11)(q23;p15) | [52] | |
| NUP98-KDM5A | t(11;12)(p15;p13) | [33] | |
| NUP98-PHF23 | t(11;17)(p15;p13) | [33] | |
| NUP98-PRRX1 | t(1;11)(q24;p15) | [33] | |
| NUP98-DDX10 | inv(11)(p15q22) | [33] | |
| MYST3-CREBBP | t(8;16)(p11;p13) | [53] | |
| RUNX1-EVI1 | t(3;21)(q26;q22) | [54] | |
| CDX2-ETV6 | t(12;13)(p13;q12) | [55] | |
| CALM-AF10 | t(10;11)(p12-14;q14-21) | [56] | |
| SET-NUP214 | del(9)(q34.11;q34.13) | [57] | |
| NPM1-MLF1 | t(3;5)(q25;q34) | [58][59] | |
| +8 | / | [60] | |
| Mutations | NPM1 | [61][62][63][64] | |
| MLL-PTD | [42] | ||
| DNMT3A | [65] | ||
| EZH2 | [42] | ||
| IDH1/2 | [63][66] | ||
| Polymorphism | GFI1-S36N | [67] |
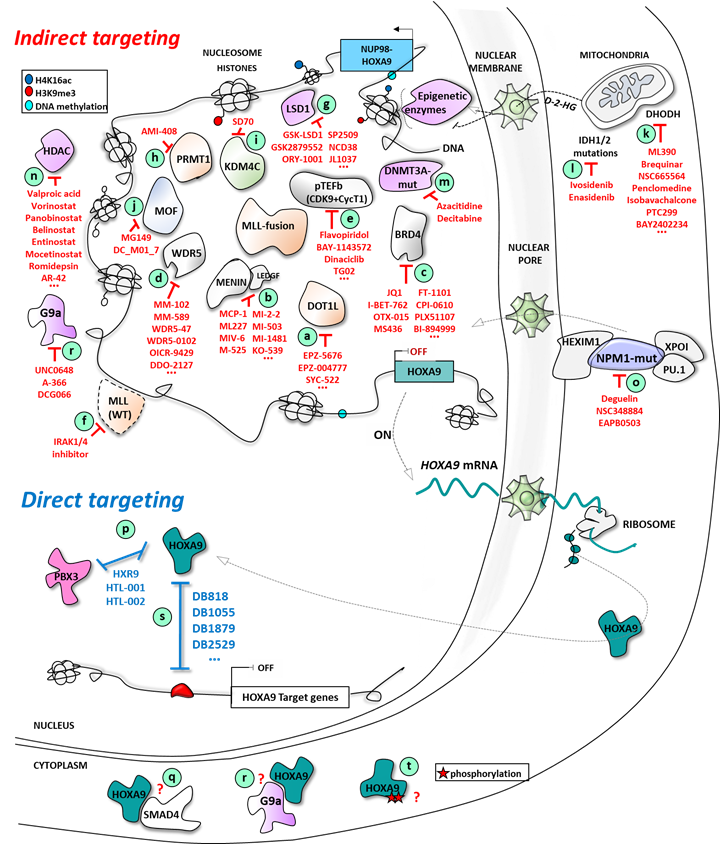
-
inhibition of its expression;
-
blockade of the specific protein/protein interaction crucial for its mechanism of action;
-
or, more specifically as part of a transcription factor, the blockade of the interaction with its cognate sequence on the DNA.
This entry is adapted from the peer-reviewed paper 10.3390/cancers11060837
References
- Vaquerizas, J.M.; Kummerfeld, S.K.; Teichmann, S.A.; Luscombe, N.M. A census of human transcription factors: Function, expression and evolution. Nat. Rev. Genet. 2009, 10, 252–263.
- Lambert, M.; Jambon, S.; Depauw, S.; David-Cordonnier, M.-H. Targeting transcription factors for cancer treatment. Molecules 2018, 23, 1479.
- Vaquerizas, J.M.; Akhtar, A.; Luscombe, N.M. Large-Scale Nuclear Architecture and Transcriptional Control. Subcell. Biochem. 2011, 52, 279–295.
- Gehring, W.J.; Affolter, M.; Burglin, T. Homeodomain Proteins—Annual Review of Biochemistry. Annu. Rev. Biochem. 1994, 63, 6487–6526.
- Lawrence, H.J.; Sauvageau, G.; Humphries, R.K.; Largman, C. The role of HOX homeobox genes in normal and leukemic hematopoiesis. Stem Cells 1996, 14, 281–291.
- Van Oostveen, J.W.; Bijl, J.J.; Raaphorst, F.M.; Walboomers, J.J.M.; Meijer, C.J.L.M. The role of Homeobox genes in normal hematopoiesis and hematological malignancies. Leukemia 1999, 13, 1675–1690.
- Argiropoulos, B.; Humphries, R.K. Hox genes in hematopoiesis and leukemogenesis. Oncogene 2007, 26, 6766–6776.
- Alharbi, R.A.; Pandha, H.S.; Simpson, G.R.; Pettengell, R.; Poterlowicz, K.; Thompson, A.; Harrington, K.; El-Tanani, M.; Morgan, R. Inhibition of HOX/PBX dimer formation leads to necroptosis in acute myeloid leukemia cells. Oncotarget 2017, 8, 89566–89579.
- Wang, K.C.; Helms, J.A.; Chang, H.Y. Regeneration, repair and remembering identity: The three Rs of Hox gene expression. Trends Cell Biol. 2009, 19, 268–275.
- Kachgal, S.; Mace, K.A.; Boudreau, N.J. The dual roles of homeobox genes in vascularization and wound healing. Cell Adhes. Migr. 2012, 6, 457–470.
- Taylor, H.S. The Role of HOX Genes in the Development and Function of the Female Reproductive Tract. Semin. Reprod. Med. 2002, 18, 081–090.
- Akbas, G.E.; Taylor, H.S. HOXC and HOXD Gene Expression in Human Endometrium: Lack of Redundancy with HOXA Paralogs1. Biol. Reprod. 2004, 70, 39–45.
- Hughes, I.A.; Acerini, C.L. Factors controlling testis descent. Eur. J. Endocrinol. 2008, 159, S75–S82.
- Raines, A.M.; Adam, M.; Magella, B.; Meyer, S.E.; Grimes, H.L.; Dey, S.K.; Potter, S.S. Recombineering-based dissection of flanking and paralogous Hox gene functions in mouse reproductive tracts. Development 2013, 140, 2942–2952.
- Du, H.; Taylor, H.S. The role of hox genes in female reproductive tract development, adult function, and fertility. Cold Spring Harb. Perspect. Med. 2016, 6, 023002.
- Awgulewitsch, A. Hox in hair growth and development. Naturwissenschaften 2003, 90, 193–211.
- Freschi, G.; Taddei, A.; Bechi, P.; Faiella, A.; Gulisano, M.; Cillo, C.; Bucciarelli, G.; Boncinelli, E. Expression of HOX homeobox genes in the adult human colonic mucosa (and colorectal cancer?). Int. J. Mol. Med. 2005, 16, 581–587.
- Mahdipour, E.; Mace, K.A. Hox transcription factor regulation of adult bone-marrow-derived cell behaviour during tissue repair and regeneration. Expert Opin. Biol. Ther. 2011, 11, 1079–1090.
- Rux, D.R.; Wellik, D.M. Hox genes in the adult skeleton: Novel functions beyond embryonic development. Dev. Dyn. 2017, 246, 310–317.
- Dunwell, T.L.; Holland, P.W.H. Diversity of human and mouse homeobox gene expression in development and adult tissues. BMC Dev. Biol. 2016, 16, 1–9.
- Pineault, N.; Helgason, C.D.; Lawrence, H.J.; Humphries, R.K. Differential expression of Hox, Meis1, and Pbx1 genes in primitive cells throughout murine hematopoietic ontogeny. Exp. Hematol. 2002, 30, 49–57.
- Barba, P.; Magli, M.C.; Tiberio, C.C.C. HOX gene expression in human cancers. Adv. Exp. Med. Biol. 1993, 348, 45–57.
- Cillo, C. HOX genes in human cancers. Invasion Metastasis 1994, 14, 38–49.
- Goodman, F.R.; Scambler, P.J. Human HOX gene mutations. Clin. Genet. 2001, 59, 1–11.
- Cantile, M.; Franco, R.; Schiavo, G.; Procino, A.; Cindolo, L.; Botti, G. The HOX genes network in uro-genital cancers: Mechanisms and potential therapeutic implications. Curr. Med. Chem. 2011, 18, 4872–4884.
- Morgan, R.; El-Tanani, M. HOX Genes as Potential Markers of Circulating Tumour Cells. Curr. Mol. Med. 2016, 16, 322–327.
- Bhatlekar, S.; Fields, J.Z.; Boman, B.M. Role of HOX Genes in Stem Cell Differentiation and Cancer. Stem Cells Int. 2018, 2018, 1–15.
- Li, B.; Huang, Q.; Wei, G.-H. The Role of HOX Transcription Factors in Cancer Predisposition and Progression. Cancers 2019, 11, 528.
- Li, Z.; Chen, P.; Su, R.; Hu, C.; Li, Y.; Elkahloun, A.G.; Zuo, Z.; Gurbuxani, S.; Arnovitz, S.; Weng, H.; et al. PBX3 and MEIS1 Cooperate in Hematopoietic Cells to Drive Acute Myeloid Leukemias Characterized by a Core Transcriptome of the MLL-Rearranged Disease. Cancer Res. 2016, 76, 619–629.
- Nakamura, T.; Largaespada, D.A.; Lee, M.P.; Johnson, L.A.; Ohyashiki, K.; Toyama, K.; Chen, S.J.; Willman, C.L.; Chen, I.M.; Feinberg, A.P.; et al. Fusion of the nucleoporin gene NUP98 to HOXA9 by the chromosome translocation t(7;11)(p15;p15) in human myeloid leukaemia. Nat. Genet. 1996, 12, 154–158.
- Kroon, E.; Krosl, J.; Thorsteinsdottir, U.; Baban, S.; Buchberg, A.M.; Sauvageau, G. Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J. 1998, 17, 3714–3725.
- Borrow, J.; Shearman, A.M.; Stanton, V.P.; Becher, R.; Collins, T.; Williams, A.J.; Dubé, I.; Katz, F.; Kwong, Y.L.; Morris, C.; et al. The t(7;11)(p15;p15) translocation in acute myeloid leukaemia fuses the genes for nucleoporin NUP98 and class I homeoprotein HOXA9. Nat. Genet. 1996, 12, 159–167.
- Gough, S.M.; Slape, C.I.; Aplan, P.D. NUP98 gene fusions and hematopoietic malignancies: Common themes and new biologic insights. Blood 2011, 118, 6247–6257.
- Rio-Machin, A.; Gómez-López, G.; Muñoz, J.; Garcia-Martinez, F.; Maiques-Diaz, A.; Alvarez, S.; Salgado, R.N.; Shrestha, M.; Torres-Ruiz, R.; Haferlach, C.; et al. The molecular pathogenesis of the NUP98-HOXA9 fusion protein in acute myeloid leukemia. Leukemia 2017, 32, 2000–2005.
- Kasper, L.H.; Brindle, P.K.; Schnabel, C.A.; Pritchard, C.E.J.; Cleary, M.L.; van Deursen, J.M.A. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol. Cell. Biol. 1999, 19, 764–776.
- Kroon, E.; Thorsteinsdottir, U.; Mayotte, N.; Nakamura, T.; Sauvageau, G. NUP98-HOXA9 expression in hemopoietic stem cells induces chronic and acute myeloid leukemias in mice. EMBO J. 2001, 20, 350–361.
- Zhang, Y.; Morrone, G.; Zhang, J.; Chen, X.; Lu, X.; Ma, L.; Moore, M.; Zhou, P. CUL-4A stimulates ubiquitylation and degradation of the HOXA9 homeodomain protein. EMBO J. 2003, 22, 6057–6067.
- Golub, T.R.; Slonim, D.K.; Tamayo, P.; Huard, C.; Gaasenbeek, M.; Mesirov, J.P.; Coller, H.; Loh, M.L.; Downing, J.R.; Caligiuri, M.A.; et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science 1999, 286, 531–537.
- Debernardi, S.; Lillington, D.M.; Chaplin, T.; Tomlinson, S.; Amess, J.; Rohatiner, A.; Lister, T.A.; Young, B.D. Genome-wide analysis of acute myeloid leukemia with normal karyotype reveals a unique pattern of homeobox gene expression distinct from those with translocation-mediated fusion events. Genes Chromosom. 2003, 37, 149–158.
- Andreeff, M.; Ruvolo, V.; Gadgil, S.; Zeng, C.; Coombes, K.; Chen, W.; Kornblau, S.; Barón, A.E.; Drabkin, H.A. HOX expression patterns identify a common signature for favorable AML. Leukemia 2008, 22, 2041–2047.
- Braekeleer, E.D.; Douet-Guilbert, N.; Basinko, A.; Bris, M.J.L.; Morel, F.; De Braekeleer, M. Hox gene dysregulation in acute myeloid leukemia. Futur. Oncol. 2014, 10, 475–495.
- Gao, L.; Sun, J.; Liu, F.; Zhang, H.; Ma, Y. Higher expression levels of the HOXA9 gene, closely associated with MLL-PTD and EZH2 mutations, predict inferior outcome in acute myeloid leukemia. Onco. Targets Ther. 2016, 9, 711–722.
- Kawagoe, H.; Humphries, R.K.; Blair, A.; Sutherland, H.J.; Hogge, D.D. Expression of HOX genes, HOX cofactors, and MLL in phenotypically and functionally defined subpopulations of leukemic and normal human hematopoietic cells. Leukemia 1999, 13, 687–698.
- Ferrando, A.A.; Armstrong, S.A.; Neuberg, D.S.; Sallan, S.E.; Silverman, L.B.; Korsmeyer, S.J.; Look, A.T. Gene expression signatures in MLL-rearranged T-lineage and B-precursor acute leukemias: Dominance of HOX dysregulation. Blood 2003, 102, 262–268.
- Faber, J.; Krivtsov, A.V.; Stubbs, M.C.; Wright, R.; Davis, T.N.; van den Heuvel-Eibrink, M.; Zwaan, C.M.; Kung, A.L.; Armstrong, S.A. HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood 2009, 113, 2375–2385.
- Wang, G.G.; Cai, L.; Pasillas, M.P.; Kamps, M.P. NUP98–NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat. Cell Biol. 2007, 9, 804–812.
- Kivioja, J.L.; Lopez Martí, J.M.; Kumar, A.; Kontro, M.; Edgren, H.; Parsons, A.; Lundán, T.; Wolf, M.; Porkka, K.; Heckman, C.A. Chimeric NUP98–NSD1 transcripts from the cryptic t(5;11)(q35.2;p15.4) in adult de novo acute myeloid leukemia. Leuk. Lymphoma 2018, 59, 725–732.
- Ghannam, G.; Takeda, A.; Camarata, T.; Moore, M.A.; Viale, A.; Yaseen, N.R. The oncogene Nup98-HOXA9 induces gene transcription in myeloid cells. J. Biol. Chem. 2004, 279, 866–875.
- Palmqvist, L.; Pineault, N.; Wasslavik, C.; Humphries, R.K. Candidate genes for expansion and transformation of hematopoietic stem cells by NUP98-HOX fusion genes. PLoS ONE 2007, 2, e768.
- Taketani, T.; Taki, T.; Shibuya, N.; Kikuchi, A.; Hanada, R.; Hayashi, Y. Novel NUP98-HOXC11 fusion gene resulted from a chromosomal break within exon 1 of HOXC11 in acute myeloid leukemia with t(11;12)(p15;q13). Cancer Res. 2002, 62, 4571–4574.
- Taketani, T.; Taki, T.; Shibuya, N.; Hayashi, Y.; Taketani, T.; Ito, E.; Kitazawa, J.; Terui, K. The HOXD11 gene is fused to the NUP98 gene in acute myeloid leukemia with t(2;11)(q31;p15). Cancer Res. 2002, 62, 33–37.
- Jankovic, D.; Gorello, P.; Liu, T.; Ehret, S.; La Starza, R.; Desjobert, C.; Baty, F.; Brutsche, M.; Jayaraman, P.S.; Santoro, A.; et al. Leukemogenic mechanisms and targets of a NUP98/HHEX fusion in acute myeloid leukemia. Blood 2008, 111, 5672–5682.
- Camós, M.; Esteve, J.; Jares, P.; Colomer, D.; Rozman, M.; Villamor, N.; Costa, D.; Carrió, A.; Nomdedéu, J.; Montserrat, E.; et al. Gene expression profiling of acute myeloid leukemia with translocation t(8;16)(p11;p13) and MYST3-CREBBP rearrangement reveals a distinctive signature with a specific pattern of HOX gene expression. Cancer Res. 2006, 66, 6947–6954.
- Jin, G.; Yamazaki, Y.; Takuwa, M.; Takahara, T.; Kaneko, K.; Kuwata, T.; Miyata, S.; Nakamura, T. Trib1 and Evi1 cooperate with Hoxa and Meis1 in myeloid leukemogenesis. Blood 2007, 109, 3998–4005.
- Chase, A.; Reiter, A.; Burci, L.; Cazzaniga, G.; Biondi, A.; Pickard, J.; Roberts, I.A.; Goldman, J.M.; Cross, N.C. Fusion of ETV6 to the caudal-related homeobox gene CDX2 in acute myeloid leukemia with the t(12;13)(p13;q12). Blood 1999, 93, 1025–1031.
- Novak, R.L.; Harper, D.P.; Caudell, D.; Slape, C.; Beachy, S.H.; Aplan, P.D. Gene expression profiling and candidate gene resequencing identifies pathways and mutations important for malignant transformation caused by leukemogenic fusion genes. Exp. Hematol. 2012, 40, 1016–1027.
- Van Vlierberghe, P.; Van Grotel, M.; Tchinda, J.; Lee, C.; Beverloo, H.B.; Van Der Spek, P.J.; Stubbs, A.; Cools, J.; Nagata, K.; Fornerod, M.; et al. The recurrent SET-NUP214 fusion as a new HOXA activation mechanism in pediatric T-cell acute lymphoblastic leukemia. Blood 2008, 111, 4668–4680.
- Dumezy, F.; Preudhomme, C.; Nibourel, O.; Labis, E.; Renneville, A.; Mayeur-Rousse, C. Acute myeloid leukemia with translocation t(3;5): New molecular insights. Haematologica 2013, 98, e52–e54.
- Lim, G.; Choi, J.R.; Kim, M.J.; Kim, S.Y.; Lee, H.J.; Suh, J.T.; Yoon, H.J.; Lee, J.; Lee, S.; Lee, W.I.; et al. Detection of t(3;5) and NPM1/MLF1 rearrangement in an elderly patient with acute myeloid leukemia: Clinical and laboratory study with review of the literature. Cancer Genet. Cytogenet. 2010, 199, 101–109.
- Kok, C.H.; Brown, A.L.; Ekert, P.G.; D’Andrea, R.J. Gene expression analysis reveals HOX gene upregulation in trisomy 8 AML. Leukemia 2010, 24, 1239–1243.
- Alcalay, M.; Tiacci, E.; Bergomas, R.; Bigerna, B.; Venturini, E.; Minardi, S.P.; Meani, N.; Diverio, D.; Bernard, L.; Tizzoni, L.; et al. Acute myeloid leukemia bearing cytoplasmic nucleophosmin (NPMc+ AML) shows a distinct gene expression profile characterized by up-regulation of genes involved in stem-cell maintenance. Blood 2005, 106, 899–902.
- Mullighan, C.G.; Kennedy, A.; Zhou, X.; Radtke, I.; Phillips, L.A.; Shurtleff, S.A.; Downing, J.R. Pediatric acute myeloid leukemia with NPM1 mutations is characterized by a gene expression profile with dysregulated HOX gene expression distinct from MLL-rearranged leukemias. Leukemia 2007, 21, 2000–2009.
- Ogawara, Y.; Katsumoto, T.; Aikawa, Y.; Shima, Y.; Kagiyama, Y.; Soga, T.; Matsunaga, H.; Seki, T.; Araki, K.; Kitabayashi, I. IDH2 and NPM1 mutations cooperate to activate Hoxa9/Meis1 and Hypoxia pathways in acute myeloid Leukemia. Cancer Res. 2015, 75, 2005–2016.
- Zhang, W.; Zhao, C.; Zhao, J.; Zhu, Y.; Weng, X.; Chen, Q.; Sun, H.; Mi, J.Q.; Li, J.; Zhu, J.; et al. Inactivation of PBX3 and HOXA9 by down-regulating H3K79 methylation represses NPM1-mutated leukemic cell survival. Theranostics 2018, 8, 4359–4371.
- Koya, J.; Kataoka, K.; Sato, T.; Bando, M.; Kato, Y.; Tsuruta-Kishino, T.; Kobayashi, H.; Narukawa, K.; Miyoshi, H.; Shirahige, K.; et al. DNMT3A R882 mutants interact with polycomb proteins to block haematopoietic stem and leukaemic cell differentiation. Nat. Commun. 2016, 7, 10924.
- Chaturvedi, A.; Araujo Cruz, M.M.; Jyotsana, N.; Sharma, A.; Yun, H.; Görlich, K.; Wichmann, M.; Schwarzer, A.; Preller, M.; Thol, F.; et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood 2013, 122, 2877–2887.
- Botezatu, L.; Michel, L.C.; Helness, A.; Vadnais, C.; Makishima, H.; Hönes, J.M.; Robert, F.; Vassen, L.; Thivakaran, A.; Al-Matary, Y.; et al. Epigenetic therapy as a novel approach for GFI136N-associated murine/human AML. Exp. Hematol. 2016, 44, 713–726.
- Rozovskaia, T.; Feinstein, E.; Mor, O.; Foa, R.; Blechman, J.; Nakamura, T.; Croce, C.M.; Cimino, G.; Canaani, E. Upregulation of Meis1 and HoxA9 in acute lymphocytic leukemias with the t(4: 11) abnormality. Oncogene 2001, 20, 874–878.
- Ayton, P.M.; Cleary, M.L. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev. 2003, 17, 2298–2307.
- Slany, R.K. The molecular biology of mixed lineage leukemia. Haematologica 2009, 94, 984–993.
- Meyer, C.; Kowarz, E.; Hofmann, J.; Renneville, A.; Zuna, J.; Trka, J.; Ben Abdelali, R.; Macintyre, E.; De Braekeleer, E.; De Braekeleer, M.; et al. New insights to the MLL recombinome of acute leukemias. Leukemia 2009, 23, 1490–1499.
- Meyer, C.; Burmeister, T.; Gröger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-De-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284.
- Marschalek, R. Mechanisms of leukemogenesis by MLL fusion proteins. Br. J. Haematol. 2011, 152, 141–154.
- Muntean, A.G.; Hess, J.L. The Pathogenesis of Mixed-Lineage Leukemia. Annu. Rev. Pathol. Mech. Dis. 2011, 7, 283–301.
- Muntean, A.G.; Tan, J.; Sitwala, K.; Huang, Y.; Bronstein, J.; Connelly, J.A.; Basrur, V.; Elenitoba-Johnson, K.S.J.; Hess, J.L. The PAF Complex Synergizes with MLL Fusion Proteins at HOX Loci to Promote Leukemogenesis. Cancer Cell 2010, 17, 609–621.
- Nguyen, T.H.; Rossetti, G.; Arnesano, F.; Ippoliti, E.; Natile, G.; Carloni, P. Molecular recognition of platinated DNA from chromosomal HMGB1. J. Chem. Theory Comput. 2014, 10, 3578–3584.
- Okuda, H.; Stanojevic, B.; Kanai, A.; Kawamura, T.; Takahashi, S.; Matsui, H.; Takaori-Kondo, A.; Yokoyama, A. Cooperative gene activation by AF4 and DOT1L drives MLL-rearranged leukemia. J. Clin. Investig. 2017, 127, 1918–1931.
- Wood, K.; Tellier, M.; Murphy, S. DOT1L and H3K79 Methylation in Transcription and Genomic Stability. Biomolecules 2018, 8, 11.
- Price, D.H. P-TEFb, a Cyclin-Dependent Kinase Controlling Elongation by RNA Polymerase II. Mol. Cell. Biol. 2002, 20, 2629–2634.
- Yokoyama, A.; Cleary, M.L. Menin Critically Links MLL Proteins with LEDGF on Cancer-Associated Target Genes. Cancer Cell 2008, 14, 36–46.
- Thiel, A.T.; Huang, J.; Lei, M.; Hua, X. Menin as a hub controlling mixed lineage leukemia. BioEssays 2012, 34, 771–780.
- El Ashkar, S.; Schwaller, J.; Pieters, T.; Goossens, S.; Demeulemeester, J.; Christ, F.; Van Belle, S.; Juge, S.; Boeckx, N.; Engelman, A.; et al. LEDGF/p75 is dispensable for hematopoiesis but essential for MLL-rearranged leukemogenesis. Blood 2018, 131, 95–107.
- Ge, Z.; Song, E.J.; Kawasawa, Y.I.; Li, J.; Dovat, S.; Song, C.; Ge, Z.; Song, E.J.; Kawasawa, Y.I.; Li, J.; et al. WDR5 high expression and its effect on tumorigenesis in leukemia. Oncotarget 2016, 7, 37740–37754.
- Deshpande, A.J.; Chen, L.; Fazio, M.; Sinha, A.U.; Bernt, K.M.; Banka, D.; Dias, S.; Chang, J.; Olhava, E.J.; Daigle, S.R.; et al. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood 2013, 121, 2533–2541.
- Ahmad, K.; Katryniok, C.; Scholz, B.; Merkens, J.; Löscher, D.; Marschalek, R.; Steinhilber, D. Inhibition of class I HDACs abrogates the dominant effect of MLL-AF4 by activation of wild-type MLL. Oncogenesis 2014, 3, e127.
- Matthews, G.M.; Mehdipour, P.; Cluse, L.A.; Falkenberg, K.J.; Wang, E.; Roth, M.; Santoro, F.; Vidacs, E.; Stanley, K.; House, C.M.; et al. Functional-genetic dissection of HDAC dependencies in mouse lymphoid and myeloid malignancies. Blood 2015, 126, 2392–2403.
- Cheung, N.; Fung, T.K.; Zeisig, B.B.; Holmes, K.; Rane, J.K.; Mowen, K.A.; Finn, M.G.; Lenhard, B.; Chan, L.C.; So, C.W.E. Targeting Aberrant Epigenetic Networks Mediated by PRMT1 and KDM4C in Acute Myeloid Leukemia. Cancer Cell 2016, 29, 32–48.
- Yu, B.D.; Hess, J.L.; Horning, S.E.; Brown, G.A.J.; Korsmeyer, S.J. Altered Hox expression and segmental identity in Mll-mutant mice. Nature 1995, 378, 505–508.
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006, 442, 818–822.
- He, M.; Chen, P.; Arnovitz, S.; Li, Y.; Huang, H.; Neilly, M.B.; Wei, M.; Rowley, J.D.; Chen, J.; Li, Z. Two isoforms of HOXA9 function differently but work synergistically in human MLL-rearranged leukemia. Blood Cells Mol. Dis. 2012, 49, 102–106.
- Zorko, N.A.; Bernot, K.M.; Whitman, S.P.; Siebenaler, R.F.; Ahmed, E.H.; Marcucci, G.G.; Yanes, D.A.; McConnell, K.K.; Mao, C.; Kalu, C.; et al. Mll partial tandem duplication and Flt3 internal tandem duplication in a double knock-in mouse recapitulates features of counterpart human acute myeloid leukemias. Blood 2012, 120, 1130–1136.
- Whitman, S.P.; Liu, S.; Vukosavljevic, T.; Rush, L.J.; Yu, L.; Liu, C.; Klisovic, M.I.; Maharry, K.; Guimond, M.; Strout, M.P.; et al. The MLL partial tandem duplication: Evidence for recessive gain-of-function in acute myeloid leukemia identifies a novel patient subgroup for molecular-targeted therapy. Blood 2005, 106, 345–352.
- Kühn, M.W.M.; Song, E.; Feng, Z.; Sinha, A.; Chen, C.W.; Deshpande, A.J.; Cusan, M.; Farnoud, N.; Mupo, A.; Grove, C.; et al. Targeting chromatin regulators inhibits leukemogenic gene expression in NPM1 mutant leukemia. Cancer Discov. 2016, 6, 1166–1181.
- Gurumurthy, M.; Tan, C.H.; Ng, R.; Zeiger, L.; Lau, J.; Lee, J.; Dey, A.; Philp, R.; Li, Q.; Lim, T.M.; et al. Nucleophosmin interacts with HEXIM1 and regulates RNA polymerase II transcription. J. Mol. Biol. 2008, 378, 302–317.
- Monroe, S.C.; Jo, S.Y.; Sanders, D.S.; Basrur, V.; Elenitoba-Johnson, K.S.; Slany, R.K.; Hess, J.L. MLL-AF9 and MLL-ENL alter the dynamic association of transcriptional regulators with genes critical for leukemia. Exp. Hematol. 2011, 39, 77–86.
- Brunetti, L.; Gundry, M.C.; Sorcini, D.; Guzman, A.G.; Huang, Y.H.; Ramabadran, R.; Gionfriddo, I.; Mezzasoma, F.; Milano, F.; Nabet, B.; et al. Mutant NPM1 Maintains the Leukemic State through HOX Expression. Cancer Cell 2018, 34, 499–512.
- Gu, X.; Ebrahem, Q.; Mahfouz, R.Z.; Hasipek, M.; Enane, F.; Radivoyevitch, T.; Rapin, N.; Przychodzen, B.; Hu, Z.; Balusu, R.; et al. Leukemogenic nucleophosmin mutation disrupts the transcription factor hub that regulates granulomonocytic fates. J. Clin. Investig. 2018, 128, 4260–4279.
- Chen, S.; Yu, J.; Lv, X.; Zhang, L. HOXA9 is critical in the proliferation, differentiation, and malignancy of leukaemia cells both in vitro and in vivo. Cell Biochem. Funct. 2017, 35, 433–440.
- Li, A.; Yang, Y.; Gao, C.; Lu, J.; Jeong, H.W.; Liu, B.H.; Tang, P.; Yao, X.; Neuberg, D.; Huang, G.; et al. A SALL4/MLL/HOXA9 pathway in murine and human myeloid leukemogenesis. J. Clin. Investig. 2013, 123, 4195–4207.
- Dickson, G.; Liberante, F.; Kettyle, L.; O’Hagan, K.; Finnegan, D.; Bullinger, L.; Geerts, D.; McMullin, M.F.; Lappin, T.; Mills, K.; et al. HOXA/PBX3 knockdown impairs growth and sensitizes cytogenetically normal acute myeloid leukemia cells to chemotherapy. Haematologica 2013, 98, 1216–1225.
- Lambert Jambon, S.; Depauw, S.; Bouhlel, M.A.; Figeac, M.; David-Cordonnier, M.-H. HOXA9 transcription factor as a target in acute myeloid leukemia: Transcription, cellular and in vivo consequences of its invalidation. Eur. J. Cancer 2016, 69, S23.
- Breitinger, C.; Maethner, E.; Garcia-Cuellar, M.P.; Slany, R.K. The homeodomain region controls the phenotype of HOX-induced murine leukemia. Blood 2012, 120, 4018–4027.
- Calvo, K.R.; Sykes, D.B.; Pasillas, M.; Kamps, M.P. Hoxa9 Immortalizes a Granulocyte-Macrophage Colony-Stimulating Factor-Dependent Promyelocyte Capable of Biphenotypic Differentiation to Neutrophils or Macrophages, Independent of Enforced Meis Expression. Mol. Cell. Biol. 2002, 20, 3274–3285.
- Hu, Y.L.; Passegué, E.; Fong, S.; Largman, C.; Lawrence, H.J. Evidence that the Pim1 kinase gene is a direct target of HOXA9. Blood 2007, 109, 4732–4738.
- Stadler, C.R.; Vegi, N.; Mulaw, M.A.; Edmaier, K.E.; Rawat, V.P.S.; Dolnik, A.; Bullinger, L.; Heilmeier, B.; Quintanilla-Fend, L.; Spiekermann, K.; et al. The leukemogenicity of Hoxa9 depends on alternative splicing. Leukemia 2014, 28, 1838–1843.
- Dintilhac, A.; Bihan, R.; Guerrier, D.; Deschamps, S.; Pellerin, I. A conserved non-homeodomain Hoxa9 isoform interacting with CBP is co-expressed with the “typical” Hoxa9 protein during embryogenesis. Gene Expr. Patterns 2004, 4, 215–222.
- Vijapurkar, U.; Fischbach, N.; Shen, W.; Brandts, C.; Stokoe, D.; Lawrence, H.J.; Largman, C. Protein kinase C-mediated phosphorylation of the leukemia-associated HOXA9 protein impairs its DNA binding ability and induces myeloid differentiation. Mol. Cell. Biol. 2004, 24, 3827–3837.
- Huang, Y.; Sitwala, K.; Bronstein, J.; Sanders, D.; Dandekar, M.; Collins, C.; Robertson, G.; MacDonald, J.; Cezard, T.; Bilenky, M.; et al. Identification and characterization of Hoxa9 binding sites in hematopoietic cells. Blood 2012, 119, 388–398.
- Zhong, X.; Prinz, A.; Steger, J.; Garcia-Cuellar, M.P.; Radsak, M.; Bentaher, A.; Slany, R.K. HoxA9 transforms murine myeloid cells by a feedback loop driving expression of key oncogenes and cell cycle control genes. Blood Adv. 2018, 2, 3137–3148.
- Dorsam, S.T.; Ferrell, C.M.; Dorsam, G.P.; Derynck, M.K.; Vijapurkar, U.; Khodabakhsh, D.; Pau, B.; Bernstein, H.; Haqq, C.M.; Largman, C.; et al. The transcriptome of the leukemogenic homeoprotein HOXA9 in human hematopoietic cells. Blood 2004, 103, 1676–1684.
- Calero-Nieto, F.J.; Joshi, A.; Bonadies, N.; Kinston, S.; Chan, W.-I.; Gudgin, E.; Pridans, C.; Landry, J.-R.; Kikuchi, J.; Huntly, B.J.; et al. HOX-mediated LMO2 expression in embryonic mesoderm is recapitulated in acute leukaemias. Oncogene 2013, 32, 5471–5480.
- Shah, C.A.; Bei, L.; Wang, H.; Platanias, L.C.; Eklund, E.A. HoxA10 Protein Regulates Transcription of Gene Encoding Fibroblast Growth Factor 2 (FGF2) in Myeloid Cells. J. Biol. Chem. 2012, 287, 18230–18248.
- Steger, J.; Füller, E.; Garcia-Cuellar, M.-P.; Hetzner, K.; Slany, R.K. Insulin-like growth factor 1 is a direct HOXA9 target important for hematopoietic transformation. Leukemia 2015, 29, 901–908.
- Brumatti, G.; Salmanidis, M.; Kok, C.H.; Bilardi, R.A.; Sandow, J.J.; Silke, N.; Manson, K.; Visser, J.; Jabbour, A.M.; Glaser, S.P.; et al. HoxA9 regulated Bcl-2 expression mediates survival of myeloid progenitors and the severity of HoxA9-dependent leukemia. Oncotarget 2013, 4, 1933–1947.
- Faderl, S.; Kantarjian, H.; Estey, E.; Manshouri, T.; Chan, C.; Rahman, E.A.; Kornblau, S.; Cortes, J.; Thomas, D.; Pierce, S.; et al. The prognostic significance of p16(INK4a)/p14(ARF) locus deletion and MDM-2 protein expression in adult acute myelogenous leukemia. Cancer 2000, 89, 1976–1982.
- Hess, J.L.; Bittner, C.B.; Zeisig, D.T.; Bach, C.; Fuchs, U.; Borkhardt, A.; Frampton, J.; Slany, R.K. c-Myb is an essential downstream target for homeobox-mediated transformation of hematopoietic cells. Blood 2006, 108, 297–304.
- Sun, Y.; Zhou, B.; Armstrong, S.A.; Dou, Y.; Hess Correspondence, J.L.; Mao, F.; Xu, J.; Miao, H.; Zou, Z.; Tran, L.; et al. HOXA9 Reprograms the Enhancer Landscape to Promote Leukemogenesis In Brief Cancer Cell HOXA9 Reprograms the Enhancer Landscape to Promote Leukemogenesis. Cancer Cell 2018, 34.
- Moskow, J.J.; Bullrich, F.; Huebner, K.; Daar, I.O.; Buchberg, A.M. Meis1, a PBX1-related homeobox gene involved in myeloid leukemia in BXH-2 mice. Mol. Cell. Biol. 1995, 15, 5434–5443.
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 188–200.
- Thorsteinsdottir, U.; Kroon, E.; Jerome, L.; Blasi, F.; Sauvageau, G. Defining roles for HOX and MEIS1 genes in induction of acute myeloid leukemia. Mol. Cell. Biol. 2001, 21, 224–234.