Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vilim Molnar | + 4314 word(s) | 4314 | 2021-08-31 10:20:59 | | | |
| 2 | Vivi Li | Meta information modification | 4314 | 2021-09-10 09:52:18 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Molnar, V. Cytokines and Chemokines in Osteoarthritis. Encyclopedia. Available online: https://encyclopedia.pub/entry/14036 (accessed on 08 February 2026).
Molnar V. Cytokines and Chemokines in Osteoarthritis. Encyclopedia. Available at: https://encyclopedia.pub/entry/14036. Accessed February 08, 2026.
Molnar, Vilim. "Cytokines and Chemokines in Osteoarthritis" Encyclopedia, https://encyclopedia.pub/entry/14036 (accessed February 08, 2026).
Molnar, V. (2021, September 09). Cytokines and Chemokines in Osteoarthritis. In Encyclopedia. https://encyclopedia.pub/entry/14036
Molnar, Vilim. "Cytokines and Chemokines in Osteoarthritis." Encyclopedia. Web. 09 September, 2021.
Copy Citation
Osteoarthritis is a common cause of disability worldwide. Although commonly referred to as a disease of the joint cartilage, osteoarthritis affects all joint tissues equally. The pathogenesis of this degenerative process is not completely understood; however, a low-grade inflammation leading to an imbalance between anabolic and katabolic processes is a well-established factor. The complex network of cytokines regulating these processes and cell communication has a central role in the development and progression of osteoarthritis. Concentrations of both proinflammatory and anti-inflammatory cytokines were found to be altered depending on the osteoarthritis stage and activity.
osteoarthritis
cytokines
chemokines
pathogenesis
inflammation
biomarker
1. Introduction
Osteoarthritis (OA) is the most common musculoskeletal condition and the largest cause of disability in the world [1]. The knee is predominantly affected in OA. A recent study concluded that knee OA globally affects 16% of the population, more often women, and that its prevalence, due to today’s lifestyle, higher obesity rates and higher average life expectancy, is constantly increasing [2]. Although OA is often referred to as a joint disease with damage and loss of cartilage, OA is a much more diverse disease with complex pathogenesis that affects all tissues within the joint [3].
One of the most important factors in the pathogenesis of OA is a disturbed cytokine balance in favor of proinflammatory cytokines that by their action initiate a vicious cycle that leads to final effects such as damage to cartilage and other intra-articular structures by activating catabolic enzymes (matrix metalloproteinases (MMPs) and ADAMTS (a disintegrin-like and metalloproteinase with thrombospondin motif)) (Figure 1) [4]. The most important inflammatory mediators in the pathogenesis of OA are IL-1β, TNF-α and IL-6. They are activators of a plethora of different signaling pathways that activate other cytokines and pathologic processes. Part of this unstoppable process are chemokines that, stimulated by cytokines, attract inflammatory cells to the joint that further promote the secretion of inflammatory factors and disease progression [5]. The aim of this review was to describe the mechanisms of action of the most important cytokines and chemokines involved in OA pathogenesis, with emphasis on knee OA.
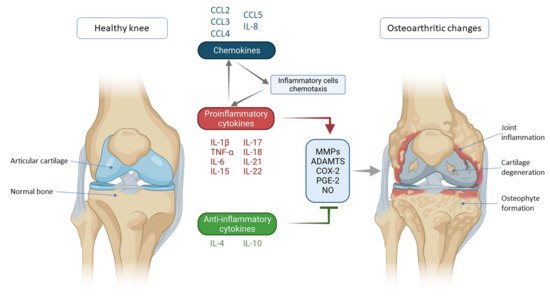
Figure 1. Schematic representation of key inflammatory processes and factors in osteoarthritis pathogenesis. The disturbed balance of proinflammatory and anti-inflammatory cytokines (in favor of proinflammatory cytokines) is responsible for the secretion of enzymes and other inflammatory factors involved in the pathogenesis of osteoarthritis leading to morphological changes within the joint such as cartilage degeneration, osteophyte formation and other inflammatory changes such as synovitis. Chemokines also contribute to inflammatory processes, stimulating the chemotaxis of inflammatory cells that then further secrete proinflammatory cytokines, thus creating a vicious circle that poses a major challenge in treating and slowing the progression of osteoarthritis. IL—interleukin; CCL-CC—chemokine ligand; TNF-α—tumor necrosis factor α; MMPs—matrix metalloproteinases (MMPs); ADAMTS—a disintegrin-like and metalloproteinase with thrombospondin motif; COX-2—cyclooxygenase-2; PGE-2—prostaglandin E2; NO—nitric oxide.
2. Cytokines and Chemokines Involved in Knee Osteoarthritis Pathogenesis
2.1. Proinflammatory Cytokines
2.1.1. IL-1β
IL-1β is one of the main proinflammatory cytokines involved in the pathogenesis of numerous diseases and a member of the IL-1 superfamily, which consists of IL-1α, IL-1β, IL36α, IL-36β, IL-36γ, IL-36RA, IL-37, IL-38 and IL-1Ra (IL-1 receptor antagonist). It achieves its effects by binding to the receptor named type I IL-1 receptor I (IL-1RI), a type I transmembrane protein that is the binding site of IL-1α and IL-1Ra as well [6]. IL-1Ra competes for an IL-1RI binding site with IL-1β with antagonistic activity. These receptors are expressed on a number of cell types in the knee joint, including chondrocytes, synoviocytes, osteoblasts, osteoclasts and inflammatory cells such as macrophages [7]. Furthermore, it has been observed that the number of IL-1RI is increased in isolated human OA chondrocytes in vitro [8]. By binding to the receptor, IL-1β activates several signaling pathways, which, combined, lead to the progression of OA. IL-1Ra binds to the same receptors as IL-1β and acts as its competitive antagonist, thus blocking IL-1β proinflammatory effects. Although IL-1Ra is an anti-inflammatory mediator, its plasma levels have been found to correlate with the radiological stage of symptomatic OA and its progression, regardless of risk factors such as age, sex and body mass index, confirming the idea of a constant competition of proinflammatory and anti-inflammatory factors in OA [9].
Through mitogen-activated protein kinase (MAPK) signaling, IL-1β induces catabolic events such as cartilage degradation, as the most dominant process in OA. MAPK consists of three families: extracellular signal-regulated kinases (ERKs), c-Jun N-terminal kinases (JNKs) and p38 MAPKs. By downregulating type II collagen and aggrecan gene expression, ERK activation by IL-1β reduces cartilage extracellular matrix (ECM) production [10]. JNK signaling also inhibits collagen synthesis through SOX-9 suppression [11]. Furthermore, IL-1β leads to ECM degradation by inducing collagenases and aggrecanases such as MMP-1 (via ERK, p38, JNK), MMP-3 (via ERK), MMP-13 (via ERK, p38, JNK), ADAMTS-4 (via ERK, p38, JNK) and ADAMTS-5 (via JNK) [12]. These catabolic events result in chondrocyte hypertrophy, dedifferentiation and, finally, apoptosis [13]. Through all three MAPK signaling pathways, IL-1β stimulates the secretion of IL-6, LIF and other proinflammatory cytokines, which potentiate the catabolic effects of IL-1β and at the same time serve as catabolic mediators on their own [12]. In that way, IL-1β can upregulate itself through a positive feedback mechanism. ERK-mediated effects can also be activated by PGE-2 (prostaglandin E2), NO (nitric oxide) and COX-2 (cyclooxygenase-2), inflammatory mediators that are, again, induced by IL-1β [14]. These mediators also contribute to synovial inflammation, which additionally enhances the secretion of IL-1β and other cytokines and aggravates the vicious circle of OA progression [15].
Another important signaling pathway in IL-1β mediated OA progression is NF-κB, which, when activated, leads to inhibition of type II collagen expression, increased production of matrix metalloproteinases (MMP-1, MMP-2, MMP-3, MMP-7, MMP-8, MMP-9 and MMP-13) and aggrecanases (ADAMTS4 and ADAMTS5), but also COX-2, iNOS, PGE-2 and NO [16][17]. Additionally, the IL-1β-activated NF-κB pathway supports proinflammatory cytokines synthesis and secretion, such as IL-6 and TNF-α [16].
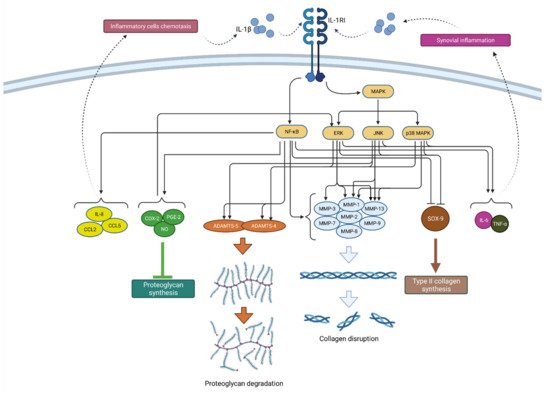
Furthermore, IL-1β-mediated NF-κB activation stimulates the production of various chemokines including IL-8, monocyte chemoattractant protein-1 (MCP-1 or CCL2), CCL5, also known as RANTES (regulated on activation, normal T cell expressed and secreted) and macrophage inflammatory protein-1a (MIP-1a), which, by attracting additional inflammatory cells, potentiate the inflammatory state in the joint [4]. In addition, activated macrophages, attracted to the synovial tissue due to the effects of chemokines, are the primary source of IL-1β secretion in the synovium, which once again confirms the complexity of the vicious inflammatory cycle in OA [12]. A schematic representation of the mechanism of action and effects of IL-1β is shown in Figure 2.
Figure 2. Schematic representation of IL-1β function in osteoarthritis pathogenesis. By binding to its receptor (IL-1RI), IL-1β activates signaling pathways (NF-κB and MAPK) that, by raising the expression of enzymes (ADAMTS and MMPs), lead to catabolic reactions, i.e., proteoglycan degradation and collagen disruption. Furthermore, via the same signaling pathways, IL-1β inhibits type II collagen synthesis through SOX-9 suppression but also proteoglycan synthesis by increasing the synthesis of COX-2, PGE-2 and NO. In addition, IL-1β increases the expression of chemokines such as IL-8, CCL2 and CCL5, as well as the cytokines IL-6 and TNF-α, which attract inflammatory cells and cause synovial inflammation, respectively, resulting in the even greater production and secretion of IL-1β. IL-1β—interleukin 1β; IL-1RI—interleukin 1 receptor 1; MAPK—mitogen-activated protein kinase; ERK—extracellular signal-regulated kinases; JNK—c-Jun N-terminal kinases; NF-κB—nuclear factor kappa-light-chain-enhancer of activated B cells; MMPs—matrix metalloproteinases (MMPs); ADAMTS—a disintegrin-like and metalloproteinase with thrombospondin motif; COX-2—cyclooxygenase-2; PGE—prostaglandin E2; NO—nitric oxide; IL-8—interleukin 8; CCL2—chemokine ligand 2; CCL—chemokine ligand 5; SOX-9—SRY-Box Transcription Factor 9; IL-6—interleukin 6; TNF-α—tumor necrosis factor α.
Due to its significant proinflammatory effects and ability to activate a number of signaling pathways in the pathogenesis of OA, the suppression of IL-1β action has been studied as a potential therapeutic method in treating OA and stopping its progression. However, IL-1β inhibition did not produce the desired effects of preventing OA progression; therefore, the negative results led to the idea that IL-1β does not likely drive OA progression [18][19][20][21]. With that in mind, researchers should consider that the pathogenesis of OA does not depend on a single cytokine; rather, the same signaling pathways can be activated by different cytokines, and the interplay of multiple factors is crucial in the onset and progression of the disease.
2.1.2. TNF-α
TNF-α is a potent proinflammatory cytokine that plays an important role in the inflammatory response. As such, it is involved in cell differentiation, proliferation and apoptosis [22]. TNF-α was discovered in 1975 by Carswell et al. as a protein that showed cytotoxic activity and caused the necrotic regression of certain tumor types. Alongside IL-1β, this cytokine is considered the key proinflammatory cytokine in the pathogenesis of OA [23].
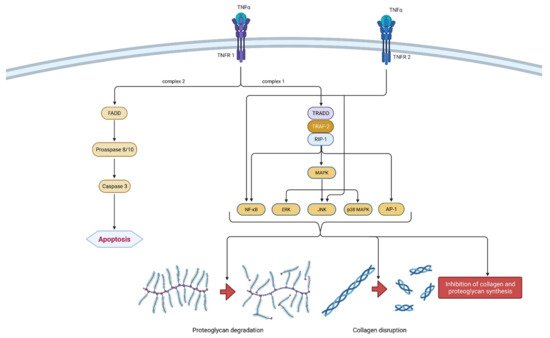
It is a part of the tumor necrosis factors superfamily, together with 18 other ligands [24]. The TNF superfamily members are type II transmembrane proteins that can be expressed in soluble and membrane-bound forms [25]. TNF-α is a homotrimeric, cone-shaped protein secreted in two forms, as mentioned above. The membrane-bound form (tmTNF-α) differs from the soluble form (sTNF-α) in its biological activity and is considered more active [26]. TNF-α binds to two isotypes of membrane receptors present on almost all known cell types except erythrocytes and unstimulated T lymphocytes. Tumor necrosis factor receptor 1 (TNRF-1) can be activated by both TNF-α forms, while TNRF-2 is mainly activated by the membrane form. Westacott et al. claim that TNRF-1 activity has a greater impact on local cartilaginous tissue loss, but both receptors are involved in signal transduction related to the pathogenesis of OA [27]. Due to their differences and unique structural features, both receptors are able to participate in different signal pathways [28]. Ligands can induce two different signaling complexes by binding to TNRF-1 receptors. Complex 1 leads to the stimulation of cell survival and the expression of pro-inflammatory genes and complex 2 leads to apoptosis and cell death. Complex 1 is associated with TNFR-1 associated death domain protein (TRADD), which allows for the binding of another two adapter proteins—receptor interacting protein-1 (RIP-1) and TNF receptor-associated factor-2 (TRAF-2). The most important transcription pathways are NF-κB and AP-1. Furthermore, another important signaling pathway is activated by mitogen-activated protein kinases (MAPK), more precisely by its three independent pathways (ERK, JNK and p38 MAPK). On the contrary, signaling complex 2 is directed towards cell death or apoptosis [28][29]. The formation of FADD (Fas-associated death domain protein), procaspase 8/10 and caspase 3 are responsible for programmed cell death. Not so long ago, TNRF-2 initiated signaling was considered less investigated than those initiated by the activation of TNRF-1 receptors. It is claimed that TNRF-2 stimulation notably supports cell activation, migration and proliferation [29]. It activates the JNK kinase and the transcription factor NF-κB. It is worth mentioning that polymorphism in the gene (M196R) encoding TNFR-2 may predetermine the development of OA by increasing the number of receptor proteins on the surface of chondrocytes [28]. The mechanism of action of TNF-α is shown in Figure 3.
Figure 3. Schematic representation of TNF-α function in osteoarthritis pathogenesis. TNF-α can bind to two receptors, TNRF-1 and TNRF-2. By binding to TNRF-1, TNF-α can induce two different signaling complexes. Complex 1 leads to the stimulation of cell survival and the expression of NF-κB, MAPK and AP-1, which results in proteoglycan degradation, collagen disruption and the inhibition of proteoglycan and collagen synthesis. On the other hand, the activation of complex 2 leads to a cascade of reactions, which include the formation of FADD and the activation of procaspase 8/10 and caspase 3, which consequently leads to cell apoptosis. Additionally, the binding of TNF-α to TNRF-2 activates NF-κB and JNK. In summation, TNF-α leads to degeneration of cartilage and other joint structures, thus contributing to the onset and progression of osteoarthritis. TNF-α—tumor necrosis factor α; TNRF-1—Tumor necrosis factor receptor 1; TNRF-2—Tumor necrosis factor receptor 2; TRADD—TNFR-1 associated death domain protein; RIP-1—receptor interacting protein-1; TRAF-2—TNF receptor-associated factor-2; MAPK—mitogen-activated protein kinase; ERK—extracellular signal-regulated kinases; JNK—c-Jun N-terminal kinases; NF-κB—nuclear factor kappa-light-chain-enhancer of activated B cells; AP-1—activator protein 1; FADD—Fas-associated death domain protein.
The activation of the same signaling pathways as IL-1ß results in synergism between these two cytokines [30]. Chondrocytes’ synthesis of proteoglycan components and type II collagen is blocked by TNF-α [31]. TNF-α also leads to extracellular matrix (ECM) degradation by inducing collagenases and aggrecanases including MMP-1, MMP-3, MMP-13 and ADAMTS-4, which coincides with IL-1β [32]. The possibility of cartilage repair is vastly reduced because of the earlier mentioned complex 2 signaling pathway and consequent cell apoptosis. Furthermore, TNF-α increases the synthesis of IL-6, IL-8, RANTES and VEGF. Together with the already mentioned IL-1β, TNF-α induces the production of iNOS, COX-2 and PGE-2 synthase, which further upregulates IL-1β and TNF-α production [28]. Considering its proinflammatory nature, it is important to mention that the inhibition of TNF-α could be a sufficient therapeutic option in treating OA. Present data suggest that monoclonal antibodies may exhibit a favorable risk-benefit ratio considering future targeted therapeutic methods. However, current monoclonal antibodies targeting TNF-α such as adalimumab, infliximab and etanercept have shown poor results in clinical studies of general OA patients. They demonstrated only limited benefits in pain reduction and no significant disease modification [33].
2.2. Anti-Inflammatory Cytokines
2.2.1. IL-4
IL-4 is a potent regulator of the immune system and is often called the prototypic immunoregulatory cytokine. It is secreted by Th2 cells, eosinophils, basophils and mast cells [34]. IL-4 is a protein consisting of 129 amino acids, and it takes the form of a four-helix bundle [28].
Its biological effect is achieved by binding to a multimeric receptor system shared with some other cytokines, such as IL-2 and IL-13. There are two different receptor type complexes. Type 1 complex is formed by the dimerization of IL-4Rα and IL-2Rγc and enables the attachment of IL-4, while the interaction between IL-4Rα and IL-13Rα1 forms type 2 complex, which enables the attachment of both IL-4 and IL-13 [28]. The exact signaling pathway of IL-4 is still not clearly described, although there is some relevant information regarding the initial intracellular events. It is known that the gradual phosphorylation of the IL-4Rα/JAK1/STAT3/STAT6 cascade leads to the expression of several proinflammatory genes [35].
There is evidence that polymorphism within the functional candidate gene IL4R is associated with OA of the hand, knee and hip [36]. Silvestri et al. found that serum soluble interleukin-4 receptor (sIL-4R) concentration was significantly higher in all OA patients compared to the healthy control group. IL-4 concentration within the synovial fluid and synovial cells was also increased [37][38]. CD4+ T-cells were detected in the sublining layer of the synovium of patients with OA, and their number was significantly higher than that of those in the same layer of healthy control. This suggests that the production of IL-4 is primarily determined by T cells (Th2) infiltrating the synovium of the joint [39]. It is worth mentioning that IL-4 has a noticeable chondroprotective effect. It inhibits the secretion of MMPs metalloproteinases, reduces the variation in the production of proteoglycans that are visible in the course of OA and, consequently, has an inhibiting effect on the degradation of proteoglycans in the articular cartilage [40][41]. Furthermore, IL-4 alone or in combination with IL-10 protects against blood-induced cartilage damage and inhibits the apoptosis of both the chondrocytes and FLS [28][42]. Considering its chondroprotective effect and the effect on other cell lineages, it is not surprising that IL-4 decreases the synthesis of inflammatory cytokines such as IL-1β, TNF-α and IL-6 [43]. In addition, IL-4 also decreases the secretion of other inflammatory mediators such as PGE-2, COX-2, PLA2 and iNOS [28].
2.2.2. IL-10
Another cytokine with pleiotropic anti-inflammatory properties is IL-10. IL-10, structurally related to interferons, initiates its effect by binding to its receptor IL-10R—a heterodimer composed of IL-10R1 and IL-10R2 subunits. Mainly produced by immune cells, IL-10 is also synthesized by chondrocytes, where it has a role in the complex mechanism of cartilage extracellular matrix turnover [44]. Upon binding, IL-10R activates the JAK-STAT kinase intracellular pathway and stimulates the expression of genes dependent on IL-10 [28]. The end product of this stimulation is a net chondroprotective, antiapoptotic and anti-inflammatory effect caused by the stimulation of type II collagen and aggrecan synthesis, as well as the inhibition of MMP synthesis [44][45]. Alternatively, IL-10 expresses its profound anti-inflammatory properties by the stimulation of IL-1β antagonist synthesis by macrophages and the inhibition of TNFα, IL-6 and IL-12, thus opposing their proinflammatory effect [28][46]. In vitro IL-10 treatment of cartilage injury model demonstrated a chondroprotective effect and increased glycosaminoglycan content (GAG). Autologous chondrocyte implant grafts treated with IL-10 also demonstrated an improvement in chondrocyte differentiation and cartilage matrix formation [47]. A recent study observed decreased serum levels of IL-10 and the decreased IL-10/TNFα ratio in patients with high-stage knee OA (Kellgren-Lawrence 4) compared with patients with moderate knee OA (Kellgren-Lawrence 3), potentially indicating its prognostic value [48]. The therapeutic effect of physical activity is often taken as an axiom in modern medicine. A clinical study exploring the effect of physical activity on IL levels in 31 female OA patients found increased levels of IL-10 intra- and periarticularly in a 3-h post-exercise period, while IL-6 and IL-8 levels remained stable throughout, thus strengthening the recommendation of physical activity for OA patients [49]. Studies also demonstrated that physical activity promotes M” anti-inflammatory macrophage phenotype differentiation, which in turn produces IL-10 and other anti-inflammatory chemokines and helps in achieving a chondroprotective anabolic joint environment [50]. The effect of mesenchymal stem cell (MSC) therapy on M2 macrophage differentiation has been established as one of the mechanisms by which they stabilize micro-inflammation in knee OA [3][50][51]. Targeted intraarticular plasmid DNA therapy was found to be safe and effective in a canine OA study, highlighting a potential for further treatment options based on IL-10 activity in knee OA [52].
2.3. Chemokines
Chemokines, also known as chemotactic cytokines, are small molecules with the ability to induce chemotaxis in a wide variety of cells. They are best known for their effect on the trafficking and guiding of immune effector cells to sites of infection or inflammation. Their wide range of action affects the proliferation, differentiation and activation of cellular responses. Thus, chemokines play an important role in persistent and ongoing inflammation in OA joints [5].
These small (8–12 kDa) protein ligands are divided into four families based on the positioning of the N-terminal cysteine residues: C, CC, CXC and CX3C. In the CC family, the cysteine residues are adjacent to each other. On the contrary, the CXC family is characterized by the separation of the two cysteine residues by an amino acid. The vast majority of known chemokines belong to these two families. The third identified chemokine family is the C family, containing a single cysteine residue in the conserved position. Finally, in the CX3C family, cysteine residues are separated similarly to the CXC family but by three variable amino acids instead of one [53]. Chemokines achieve their effects by binding to G-protein coupled cell-surface receptors. These receptors show different levels of binding specificity and promiscuity, but they do not bind different groups of chemokines. For example, CCR receptors bind only CCL chemokine ligands and CXCR receptors bind CXCL ligands. In order to understand the importance of chemokines in the course of OA, it is inevitable to mention their role in driving cellular motility during the inflammatory response. Leukocytes express a specific set of chemokine receptors and migrate to sites of infection or tissue damage along the gradients of their cognate chemokine ligands. Furthermore, chemokines arrange the recruitment of pluripotent cell types to sites of tissue repair. They perform a variety of functions aside from chemotaxis, including T helper cell differentiation and function as well as angiogenesis, and have a pleiotropic effect on multiple cell types related to the pathogenesis of OA [5][54].
The most important CC family chemokines that are related to OA are CCL2, CCL3, CCL4 and CCL5 [5]. The monocyte chemoattractant protein-1 (MCP-1/CCL2) is a potent chemotactic factor for monocytes that also recruits memory T-lymphocytes and natural killer (NK) cells. Its effects are primarily associated with its binding to the CCR2 receptor [55]. Elevated levels of CCL2 were found in the synovial fluid of patients with both knee injuries and knee OA [56][57]. Miller et al. found that both CCL2 and CCR2 were upregulated in the innervating dorsal root ganglia (DRG) of the knee 8 weeks after surgical injury in a murine model [58]. The same authors did a follow-up study and reported that CCL2 production by murine DRG neurons was induced by alarmin S100A8 and the plasma protein α2 macroglobulin, which are molecular “danger signals” strongly involved in OA pathogenesis [59]. CCL2 (MCP-1) production is dependent on Toll-like receptor-4 (TLR-4) signaling. These findings imply that products of tissue damage and inflammation during OA could stimulate nociceptive pathways. Genetic variation in the CCL2 gene may be associated with knee OA [60]. CCL2 increases MMP-3 expression, which results in proteoglycan loss and the degradation of cartilaginous tissue [61].
CCL3 (MIP-1α), CCL4 (MIP-1β), and CCL5 (RANTES) are other members of the CC family that are also upregulated in OA. Zhao et al. investigated chemokine levels in the plasma of 181 patients (75 control patients, 47 pre-radiographic knee OA patients and 50 radiographic knee OA patients) [62]. CCL3 in plasma showed the highest ability to discriminate pre-radiographic knee OA patients from the control group. Levels in plasma increased with the radiographic severity of the disease. Beekhuizen et al. found that CCL5 levels were among the most significantly elevated mediators in OA synovial fluid compared with controls [63]. Another study that confirms this statement documented CCL5 levels elevation in 18 additional patients [56]. It is worth mentioning that all of these three chemokines are ligands for CCR5. Consequently, Takabe et al. found that CCR5 deficient mice were partially protected against post-traumatic cartilage erosion [64]. There were no signs of bone remodeling or synovial response to surgery, suggesting that CCR5 functions primarily in cartilage during the development of post-traumatic OA. IL-1β-treated human chondrocytes showed the significant upregulation of CCL3, CCL4 and CCL5 [65].
Chemokines from the CXC family that play a significant role in the pathogenesis of OA are CXCL8 (IL-8) and CXCL12. IL-8 is a chemokine molecule, first described as a chemoattractant of neutrophils. Today it is known that IL-8 exhibits effects on many different cells, and it is researched in numerous diseases [66]. It is expressed by cells of the immune system, most prominently CD8+ T cells, macrophages and monocytes, but also by keratinocytes, fibroblasts, epithelial cells, hepatocytes and synovial cells [66]. It acts on CXCR1 and CXCR2 receptors expressed not only on leukocytes but also on chondrocytes, osteoclasts, fibroblasts, epithelial and endothelial cells and on the cells of the nervous system [66][67][68].
It has been shown on the human chondrocyte cell line (CHON-002) that IL-8 can be upregulated by TNF-α [69]. Furthermore, IL-8 production is stimulated by advanced glycation end products (AGEs) through NF-κB signaling, which are known to accumulate in cartilage with age and stimulate catabolic metabolism in chondrocytes [70]. Additionally, it has been shown that in human OA chondrocytes, IL-8 is regulated by DNA demethylation that is affected by IL-1b signaling [71]. Free fatty acids also increase the production of IL-8 in the osteoblasts of patients with OA but have little effect on IL-8 secretion in osteoclasts [72]. Osteopontin is yet another molecule involved in the regulation of IL-8 expression, and it is known to stimulate IL-8 in chondrocytes [73]. The mechanical load also increases IL-8 secretion in the chondrocytes of OA patients [74].
Without a doubt, IL-8 is significantly more expressed in the synovial tissue and synovial fluid of patients with RA than in OA [75][76][77][78][79][80]. OA patients undergoing surgery had 37-fold higher IL-8 expression in chondrocytes than patients undergoing surgery due to a fracture of the neck of the femur (likely due to osteoporosis) [71]. Koh et al. have shown that IL-8 is higher in the synovial fluid of OA patients than in young patients with ligament injury [81]. This is also supported by animal studies demonstrating increased IL-8 in dogs with OA [82][83]. Furthermore, it has been shown that IL-8 is also slightly higher in the serum of OA patients than in healthy control [77][81]. IL-8 in synovial fluid has been shown to correlate with the clinical severity of OA, but IL-8 in serum has not [84]. On the other hand, Ruan et al. have demonstrated a certain correlation between serum IL-8 and the clinically and radiologically assessed severity of OA [84][85].
IL-8 is also known to increase collagen I, MMP1- and MMP-13 protein concentration and to enhance the phosphorylation of STAT3 and NF-Kb subunit p65 [69]. It can also affect chondrocyte morphology by decreasing endogenous GTP-Cdc42 and increasing stress fibers. HA concentration in the knee negatively correlates with IL-8 in synovial fluid [80]. In patients with a good response to sodium hyaluronate treatment in terms of improvement of hydrarthrosis, there was a prominent reduction of IL-8 and IL-6 concentration following the treatment [80]. IL-8 also stimulates the hypertrophy of chondrocytes and the calcifications of the matrix [67]. Further studies by the same group have shown that IL-8 increases the expression of PiT-1 expression and stimulates the uptake of inorganic phosphate in chondrocytes [86].
CXCL12, also known as stromal cell-derived factor-1 (SDF-1), is a chemokine that plays a key role in tissue regeneration. It mobilizes mesenchymal stem cells (MSCs) to sites of injury by binding to CXCR4 [87]. Shen et al. confirmed this statement by studying the effects of human meniscus-derived stem/progenitor cells (hMeSPCs) in a rat meniscectomy model [88]. hMeSPCs were injected intra-articularly after meniscectomy and homed to the injured meniscus. The meniscal repair was superior in the hMeSPCs-treated mice, with significantly reduced cartilage degeneration. In a study consisting of 252 patients with knee OA and 144 healthy controls, CXCL12 levels in the synovial fluid were closely related to the radiographic severity of OA [89]. Besides their effect on MSCs, there is evidence that articular chondrocytes express CXCR4, and CXCL12 also induces MMP13 and some other catabolic mediators. The disruption of these catabolic events could be achieved by the pharmacological blockade of CXCL2/CXCR4 signaling. Thus, the disruption of the CXCL12/CXCR4 signaling can be used as a therapeutic approach to attenuate cartilage degeneration in OA [90]. Taking into consideration all of the above, it is obvious that CXCL12 has diverse effects that depend on cellular targets.
References
- Neogi, T. The epidemiology and impact of pain in osteoarthritis. Osteoarthr. Cartil. 2013, 21, 1145–1153.
- Cui, A.; Li, H.; Wang, D.; Zhong, J.; Chen, Y.; Lu, H. Global, regional prevalence, incidence and risk factors of knee osteoarthritis in population-based studies. EClinicalMedicine 2020, 29–30, 100587.
- Primorac, D.; Molnar, V.; Rod, E.; Jeleč, Ž.; Čukelj, F.; Matišić, V.; Vrdoljak, T.; Hudetz, D.; Hajsok, H.; Borić, I. Knee Osteoarthritis: A Review of Pathogenesis and State-Of-The-Art Non-Operative Therapeutic Considerations. Genes 2020, 11, 854.
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.-P.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42.
- Scanzello, C.R. Chemokines and inflammation in osteoarthritis: Insights from patients and animal models. J. Orthop. Res. 2017, 35, 735–739.
- Fields, J.K.; Günther, S.; Sundberg, E.J. Structural Basis of IL-1 Family Cytokine Signaling. Front. Immunol. 2019, 10, 1412.
- Boraschi, D.; Italiani, P.; Weil, S.; Martin, M.U. The family of the interleukin-1 receptors. Immunol. Rev. 2018, 281, 197–232.
- Martel-Pelletier, J.; Mccollum, R.; Dibattista, J.; Faure, M.-P.; Chin, J.A.; Fournier, S.; Sarfati, M.; Pelletier, J.-P. The interleukin-1 receptor in normal and osteoarthritic human articular chondrocytes. Identification as the type I receptor and analysis of binding kinetics and biologic function. Arthritis Rheum. 1992, 35, 530–540.
- Attur, M.; Statnikov, A.; Samuels, J.; Li, Z.; Alekseyenko, A.V.; Greenberg, J.D.; Krasnokutsky, S.; Rybak, L.; Lu, Q.A.; Todd, J.; et al. Plasma levels of interleukin-1 receptor antagonist (IL1Ra) predict radiographic progression of symptomatic knee osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1915–1924.
- Wang, X.; Li, F.; Fan, C.; Wang, C.; Ruan, H. Effects and relationship of ERK1 and ERK2 in interleukin-1β-induced alterations in MMP3, MMP13, type II collagen and aggrecan expression in human chondrocytes. Int. J. Mol. Med. 2011, 27, 583–589.
- Hwang, S.-G.; Yu, S.-S.; Poo, H.; Chun, J.-S. c-Jun/Activator Protein-1 Mediates Interleukin-1β-induced Dedifferentiation but Not Cyclooxygenase-2 Expression in Articular Chondrocytes. J. Biol. Chem. 2005, 280, 29780–29787.
- Jenei-Lanzl, Z.; Meurer, A.; Zaucke, F. Interleukin-1β signaling in osteoarthritis—chondrocytes in focus. Cell. Signal. 2019, 53, 212–223.
- Hwang, H.; Kim, H. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2015, 16, 26035–26054.
- Wang, X.; Li, F.; Fan, C.; Wang, C.; Ruan, H. Analysis of isoform specific ERK signaling on the effects of interleukin-1β on COX-2 expression and PGE2 production in human chondrocytes. Biochem. Biophys. Res. Commun. 2010, 402, 23–29.
- Chow, Y.Y.; Chin, K.-Y. The Role of Inflammation in the Pathogenesis of Osteoarthritis. Mediat. Inflamm. 2020, 2020, 1–19.
- Choi, M.C.; Jo, J.; Park, J.; Kang, H.K.; Park, Y. NF-B Signaling Pathways in Osteoarthritic Cartilage Destruction. Cells 2019, 8, 734.
- Lepetsos, P.; Papavassiliou, K.A.; Papavassiliou, A.G. Redox and NF-κB signaling in osteoarthritis. Free Radic. Biol. Med. 2019, 132, 90–100.
- Chevalier, X.; Eymard, F.; Richette, P. Biologic agents in osteoarthritis: Hopes and disappointments. Nat. Rev. Rheumatol. 2013, 9, 400–410.
- Chevalier, X.; Goupille, P.; Beaulieu, A.D.; Burch, F.X.; Bensen, W.G.; Conrozier, T.; Loeuille, D.; Kivitz, A.J.; Silver, D.; Appleton, B.E. Intraarticular injection of anakinra in osteoarthritis of the knee: A multicenter, randomized, double-blind, placebo-controlled study. Arthritis Care Res. 2009, 61, 344–352.
- Chevalier, X.; Eymard, F. Anti-IL-1 for the treatment of OA: Dead or alive? Nat. Rev. Rheumatol. 2019, 15, 191–192.
- Theeuwes, W.F.; van den Bosch, M.H.J.; Thurlings, R.M.; Blom, A.B.; van Lent, P.L.E.M. The role of inflammation in mesenchymal stromal cell therapy in osteoarthritis, perspectives for post-traumatic osteoarthritis: A review. Rheumatology 2021, 60, 1042–1053.
- Baud, V.; Karin, M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001, 11, 372–377.
- Bodmer, J.L.; Schneider, P.; Tschopp, J. The molecular architecture of the TNF superfamily. Trends Biochem. Sci. 2002, 27, 19–26.
- Kriegler, M.; Perez, C.; DeFay, K.; Albert, I.; Lu, S.D. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: Ramifications for the complex physiology of TNF. Cell 1988, 53, 45–53.
- Zhou, T.; Mountz, J.D.; Kimberly, R.P. Immunobiology of tumor necrosis factor receptor superfamily. Immunol. Res. 2002, 26, 323–336.
- Appay, V.; Sauce, D. Immune activation and inflammation in HIV-1 infection: Causes and consequences. J. Pathol. 2008, 214, 231–241.
- Westacott, C.I.; Barakat, A.F.; Wood, L.; Perry, M.J.; Neison, P.; Bisbinas, I.; Armstrong, L.; Millar, A.B.; Elson, C.J. Tumor necrosis factor alpha can contribute to focal loss of cartilage in osteoarthritis. Osteoarthr. Cartil. 2000, 8, 213–221.
- Wojdasiewicz, P.; Poniatowski, Ł.A.; Szukiewicz, D. The Role of Inflammatory and Anti-Inflammatory Cytokines in the Pathogenesis of Osteoarthritis. Mediat. Inflamm. 2014, 2014, 1–19.
- Zelová, H.; Hošek, J. TNF-α signalling and inflammation: Interactions between old acquaintances. Inflamm. Res. 2013, 62, 641–651.
- Henderson, B.; Pettipher, E.R. Arthritogenic actions of recombinant IL-1 and tumour necrosis factor α in the rabbit: Evidence for synergistic interactions between cytokines in vivo. Clin. Exp. Immunol. 1989, 75, 306–310.
- Séguin, C.A.; Bernier, S.M. TNFα Suppresses Link Protein and Type II Collagen Expression in Chondrocytes: Role of MEK1/2 and NF-κB Signaling Pathways. J. Cell. Physiol. 2003, 197, 356–369.
- Xue, J.; Wang, J.; Liu, Q.; Luo, A. Tumor necrosis factor-α induces ADAMTS-4 expression in human osteoarthritis chondrocytes. Mol. Med. Rep. 2013, 8, 1755–1760.
- Oo, W.M.; Yu, S.P.-C.; Daniel, M.S.; Hunter, D.J. Disease-modifying drugs in osteoarthritis: Current understanding and future therapeutics. Expert Opin. Emerg. Drugs 2018, 23, 331–347.
- Brown, M.A.; Hural, J. Functions of IL-4 and control of its expression. Crit. Rev. Immunol. 2017, 37, 181–212.
- Bhattacharjee, A.; Shukla, M.; Yakubenko, V.P.; Mulya, A.; Kundu, S.; Cathcart, M.K. IL-4 and IL-13 employ discrete signaling pathways for target gene expression in alternatively activated monocytes/macrophages. Free Radic. Biol. Med. 2013, 54, 1–16.
- Forster, T.; Chapman, K.; Loughlin, J. Common variants within the interleukin 4 receptor α gene (IL4R) are associated with susceptibility to osteoarthritis. Hum. Genet. 2004, 114, 391–395.
- Schlaak, J.F.; Pfers, I.; Meyer Zum Büschenfelde, K.H.; Märker-Hermann, E. Different cytokine profiles in the synovial fluid of patients with osteoarthritis, rheumatoid arthritis and seronegative spondylarthropathies. Clin. Exp. Rheumatol. 1996, 14, 155–162.
- Wagner, S.; Fritz, P.; Einsele, H.; Sell, S.; Saal, J.G. Evaluation of synovial cytokine patterns in rheumatoid arthritis and osteoarthritis by quantitative reverse transcription polymerase chain reaction. Rheumatol. Int. 1997, 16, 191–196.
- Ishii, H.; Tanaka, H.; Katoh, K.; Nakamura, H.; Nagashima, M.; Yoshino, S. Characterization of infiltrating T cells and Th1/Th2-type cytokines in the synovium of patients with osteoarthritis. Osteoarthr. Cartil. 2002, 10, 277–281.
- Yeh, L.A.; Augustine, A.J.; Lee, P.; Riviere, L.R.; Sheldon, A. Interleukin-4, an inhibitor of cartilage breakdown in bovine articular cartilage explants. J. Rheumatol. 1995, 22, 1740–1746.
- Doi, H.; Nishida, K.; Yorimitsu, M.; Komiyama, T.; Kadota, Y.; Tetsunaga, T.; Yoshida, A.; Kubota, S.; Takigawa, M.; Ozaki, T. Interleukin-4 downregulates the cyclic tensile stress-induced matrix metalloproteinases-13 and cathepsin B expression by rat normal chondrocytes. Acta Med. Okayama 2008, 62, 119–126.
- Van Meegeren, M.E.R.; Roosendaal, G.; Jansen, N.W.D.; Wenting, M.J.G.; Van Wesel, A.C.W.; Van Roon, J.A.G.; Lafeber, F.P.J.G. IL-4 alone and in combination with IL-10 protects against blood-induced cartilage damage. Osteoarthr. Cartil. 2012, 20, 764–772.
- Schuerwegh, A.J.; Dombrecht, E.J.; Stevens, W.J.; Van Offel, J.F.; Bridts, C.H.; De Clerck, L.S. Influence of pro-inflammatory (IL-1α, IL-6, TNF-α, IFN-γ) and anti-inflammatory (IL-4) cytokines on chondrocyte function. Osteoarthr. Cartil. 2003, 11, 681–687.
- Schulze-Tanzil, G.; Zreiqat, H.; Sabat, R.; Kohl, B.; Halder, A.; Muller, R.; John, T. Interleukin-10 and Articular Cartilage: Experimental Therapeutical Approaches in Cartilage Disorders. Curr. Gene Ther. 2009, 9, 306–315.
- Mostafa, E.; Chollet-martin, S.; Oudghiri, M.; Laquay, N.; Jacob, M.; Michel, J.; Feldman, L.J. Effects of interleukin-10 on monocyte / endothelial cell adhesion and MMP-9/TIMP-1 secretion. Cardiovasc. Res. 2001, 49, 882–890.
- Wang, Y.S.; Wang, Y.H.; Zhao, G.Q.; Li, Y.B. Osteogenic potential of human calcitonin gene-related peptide alpha gene-modified bone marrow mesenchymal stem cells. Chin. Med. J. 2011, 124, 3976–3981.
- Behrendt, P.; Feldheim, M.; Preusse-Prange, A.; Weitkamp, J.T.; Haake, M.; Eglin, D.; Rolauffs, B.; Fay, J.; Seekamp, A.; Grodzinsky, A.J.; et al. Chondrogenic potential of IL-10 in mechanically injured cartilage and cellularized collagen ACI grafts. Osteoarthr. Cartil. 2018, 26, 264–275.
- Barker, T.; Rogers, V.E.; Henriksen, V.T.; Trawick, R.H.; Momberger, N.G.; Lynn Rasmussen, G. Circulating IL-10 is compromised in patients predisposed to developing and in patients with severe knee osteoarthritis. Sci. Rep. 2021, 11, 1–10.
- Helmark, I.C.; Mikkelsen, U.R.; Børglum, J.; Rothe, A.; Petersen, M.C.; Andersen, O.; Langberg, H.; Kjaer, M. Exercise increases interleukin-10 levels both intraarticularly and peri-synovially in patients with knee osteoarthritis: A randomized controlled trial. Arthritis Res. Ther. 2010, 12, R126.
- Fernandes, T.L.; Gomoll, A.H.; Lattermann, C.; Hernandez, A.J.; Bueno, D.F.; Amano, M.T. Macrophage: A Potential Target on Cartilage Regeneration. Front. Immunol. 2020, 11, 111.
- Zhang, J.; Rong, Y.; Luo, C.; Cui, W. Bone marrow mesenchymal stem cell-derived exosomes prevent osteoarthritis by regulating synovial macrophage polarization. Aging 2020, 12, 25138–25152.
- Watkins, L.R.; Chavez, R.A.; Landry, R.; Fry, M.; Green-Fulgham, S.M.; Coulson, J.D.; Collins, S.D.; Glover, D.K.; Rieger, J.; Forsayeth, J.R. Targeted interleukin-10 plasmid DNA therapy in the treatment of osteoarthritis: Toxicology and pain efficacy assessments. Brain. Behav. Immun. 2020, 90, 155–166.
- Borish, L.C.; Steinke, J.W. 2. Cytokines and chemokines. J. Allergy Clin. Immunol. 2003, 111, S460–S475.
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta- Mol. Cell Res. 2014, 1843, 2563–2582.
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interf. Cytokine Res. 2009, 29, 313–325.
- Monibi, F.; Roller, B.L.; Stoker, A.; Garner, B.; Bal, S.; Cook, J.L. Identification of Synovial Fluid Biomarkers for Knee Osteoarthritis and Correlation with Radiographic Assessment. J. Knee Surg. 2016, 29, 242–247.
- Watt, F.E.; Paterson, E.; Freidin, A.; Kenny, M.; Judge, A.; Saklatvala, J.; Williams, A.; Vincent, T.L. Acute Molecular Changes in Synovial Fluid Following Human Knee Injury: Association With Early Clinical Outcomes. Arthritis Rheumatol. 2016, 68, 2129–2140.
- Miller, R.E.; Tran, P.B.; Das, R.; Ghoreishi-Haack, N.; Ren, D.; Miller, R.J.; Malfait, A.M. CCR2 chemokine receptor signaling mediates pain in experimental osteoarthritis. Proc. Natl. Acad. Sci. USA 2012, 109, 20602–20607.
- Miller, R.E.; Belmadani, A.; Ishihara, S.; Tran, P.B.; Ren, D.; Miller, R.J.; Malfait, A.M. Damage-associated molecular patterns generated in osteoarthritis directly excite murine nociceptive neurons through toll-like receptor 4. Arthritis Rheumatol. 2015, 67, 2933–2943.
- Hulin-Curtis, S.L.; Bidwell, J.L.; Perry, M.J. Association between CCL2 haplotypes and knee osteoarthritis. Int. J. Immunogenet. 2013, 40, 280–283.
- Borzí, R.M.; Mazzetti, I.; Cattini, L.; Uguccioni, M.; Baggiolini, M.; Facchini, A. Human chondrocytes express functional chemokine receptors and release matrix-degrading enzymes in response to C-X-C and C-C chemokines. Arthritis Rheum. 2000, 43, 1734–1741.
- Zhao, X.Y.; Yang, Z.B.; Zhang, Z.J.; Zhang, Z.Q.; Kang, Y.; Huang, G.X.; Wang, S.W.; Huang, H.; Liao, W.M. CCL3 serves as a potential plasma biomarker in knee degeneration (osteoarthritis). Osteoarthr. Cartil. 2015, 23, 1405–1411.
- Beekhuizen, M.; Gierman, L.M.M.; van Spil, W.E.E.; Van Osch, G.J.V.M.J.V.M.; Huizinga, T.W.J.W.J.; Saris, D.B.F.B.F.; Creemers, L.B.B.; Zuurmond, A.-M.M. An explorative study comparing levels of soluble mediators in control and osteoarthritic synovial fluid. Osteoarthr. Cartil. 2013, 21, 918–922.
- Takebe, K.; Rai, M.F.; Schmidt, E.J.; Sandell, L.J. The chemokine receptor CCR5 plays a role in post-traumatic cartilage loss in mice, but does not affect synovium and bone. Osteoarthr. Cartil. 2015, 23, 454–461.
- Sandell, L.J.; Xing, X.; Franz, C.; Davies, S.; Chang, L.W.; Patra, D. Exuberant expression of chemokine genes by adult human articular chondrocytes in response to IL-1β. Osteoarthr. Cartil. 2008, 16, 1560–1571.
- Russo, R.C.; Garcia, C.C.; Teixeira, M.M.; Amaral, F.A. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev. Clin. Immunol. 2014, 10, 593–619.
- Merz, D.; Liu, R.; Johnson, K.; Terkeltaub, R. IL-8/CXCL8 and Growth-Related Oncogene α/CXCL1 Induce Chondrocyte Hypertrophic Differentiation. J. Immunol. 2003, 171, 4406–4415.
- Galliera, E.; Locati, M.; Mantovani, A.; Corsi, M.M. Chemokines and bone remodeling. Int. J. Immunopathol. Pharmacol. 2008, 21, 485–491.
- Sun, F.; Zhang, Y.; Li, Q. Therapeutic mechanisms of ibuprofen, prednisone and betamethasone in osteoarthritis. Mol. Med. Rep. 2017, 15, 981–987.
- Rasheed, Z.; Akhtar, N.; Haqqi, T.M. Advanced glycation end products induce the expression of interleukin-6 and interleukin-8 by receptor for advanced glycation end product-mediated activation of mitogen-activated protein kinases and nuclear factor-κB in human osteoarthritis chondrocytes. Rheumatology 2011, 50, 838–851.
- Takahashi, A.; de Andrés, M.C.; Hashimoto, K.; Itoi, E.; Oreffo, R.O.C. Epigenetic regulation of interleukin-8, an inflammatory chemokine, in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1946–1954.
- Frommer, K.W.; Hasseli, R.; Schäffler, A.; Lange, U.; Rehart, S.; Steinmeyer, J.; Rickert, M.; Sarter, K.; Zaiss, M.M.; Culmsee, C.; et al. Free Fatty Acids in Bone Pathophysiology of Rheumatic Diseases. Front. Immunol. 2019, 10, 2757.
- Yang, Y.; Gao, S.G.; Zhang, F.J.; Luo, W.; Xue, J.X.; Lei, G.H. Effects of osteopontin on the expression of IL-6 and IL-8 inflammatory factors in human knee osteoarthritis chondrocytes. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 3580–3586.
- Sakao, K.; Takahashi, K.A.; Arai, Y.; Saito, M.; Honjo, K.; Hiraoka, N.; Asada, H.; Shin-Ya, M.; Imanishi, J.; Mazda, O.; et al. Osteoblasts derived from osteophytes produce interleukin-6, interleukin-8, and matrix metalloproteinase-13 in osteoarthritis. J. Bone Miner. Metab. 2009, 27, 412–423.
- Remick, D.G.; DeForge, L.E.; Sullivan, J.F.; Showell, H.J. Profile of cytokines in synovial fluid specimens from patients with arthritis. Immunol. Invest. 1992, 21, 321–327.
- Lee, Y.A.; Choi, H.M.; Lee, S.H.; Yang, H.I.; Yoo, M.C.; Hong, S.J.; Kim, K.S. Synergy between adiponectin and interleukin-1β on the expression of interleukin-6, interleukin-8, and cyclooxygenase-2 in fibroblast-like synoviocytes. Exp. Mol. Med. 2012, 44, 440–447.
- Furuzawa-Carballeda, J.; Alcocer-Varela, J. Interleukin-8, interleukin- 10, intercellular adhesion molecule- 1 and vascular cell adhesion molecule-1 expression levels are higher in synovial tissue from patients with rheumatoid arthritis than in osteoarthritis. Scand. J. Immunol. 1999, 50, 215–222.
- Bertazzolo, N.; Punzi, L.; Stefani, M.P.; Cesaro, G.; Pianon, M.; Finco, B.; Todesco, S. Interrelationships between interleukin (IL)-1, IL-6 and IL-8 in synovial fluid of various arthropathies. Agents Actions 1994, 41, 90–92.
- Valcamonica, E.; Chighizola, C.B.; Comi, D.; De Lucia, O.; Pisoni, L.; Murgo, A.; Salvi, V.; Sozzani, S.; Meroni, P.L. Levels of chemerin and interleukin 8 in the synovial fluid of patients with inflammatory arthritides and osteoarthritis. Clin. Exp. Rheumatol. 2014, 32, 243–250.
- Kaneko, S.; Satoh, T.; Chiba, J.; Ju, C.; Inoue, K.; Kagawa, J. Interleukin-6 and interleukin-8 levels in serum and synovial fluid of patients with osteoarthritis. Cytokines Cell. Mol. Ther. 2000, 6, 71–79.
- Koh, S.M.M.; Chan, C.K.K.; Teo, S.H.H.; Singh, S.; Merican, A.; Ng, W.M.M.; Abbas, A.; Kamarul, T. Elevated plasma and synovial fluid interleukin-8 and interleukin-18 may be associated with the pathogenesis of knee osteoarthritis. Knee 2020, 27, 26–35.
- Allen, P.I.; Conzemius, M.G.; Evans, R.B.; Kiefer, K. Correlation between synovial fluid cytokine concentrations and limb function in normal dogs and in dogs with lameness from spontaneous osteoarthritis. Vet. Surg. 2019, 48, 770–779.
- Kleine, S.A.; Gogal, R.M.; George, C.; Thaliath, M.; Budsberg, S.C. Elevated Synovial Fluid Concentration of Monocyte Chemoattractant Protein-1 and Interleukin-8 in Dogs with Osteoarthritis of the Stifle. Vet. Comp. Orthop. Traumatol. 2020, 33, 147–150.
- García-Manrique, M.; Calvet, J.; Orellana, C.; Berenguer-Llergo, A.; Garcia-Cirera, S.; Llop, M.; Albiñana-Giménez, N.; Galisteo-Lencastre, C.; Gratacós, J. Synovial fluid but not plasma interleukin-8 is associated with clinical severity and inflammatory markers in knee osteoarthritis women with joint effusion. Sci. Rep. 2021, 11, 1–7.
- Ruan, G.; Xu, J.; Wang, K.; Zheng, S.; Wu, J.; Bian, F.; Chang, B.; Zhang, Y.; Meng, T.; Zhu, Z.; et al. Associations between serum IL-8 and knee symptoms, joint structures, and cartilage or bone biomarkers in patients with knee osteoarthritis. Clin. Rheumatol. 2019, 38, 3609–3617.
- Cecil, D.L.; Rose, D.M.; Terkeltaub, R.; Liu-Bryan, R. Role of interleukin-8 in PiT-1 expression and CXCR1-mediated inorganic phosphate uptake in chondrocytes. Arthritis Rheum. 2005, 52, 144–154.
- Marquez-Curtis, L.A.; Janowska-Wieczorek, A. Enhancing the migration ability of mesenchymal stromal cells by targeting the SDF-1/CXCR4 axis. Biomed Res. Int. 2013, 2013, 1–15.
- Shen, W.; Chen, J.; Zhu, T.; Chen, L.; Zhang, W.; Fang, Z.; Heng, B.C.; Yin, Z.; Chen, X.; Ji, J.; et al. Intra-Articular Injection of Human Meniscus Stem/Progenitor Cells Promotes Meniscus Regeneration and Ameliorates Osteoarthritis Through Stromal Cell-Derived Factor-1/CXCR4-Mediated Homing. Stem Cells Transl. Med. 2014, 3, 387–394.
- Gao, F.; Tian, J.; Pan, H.; Gao, J.; Yao, M. Association of CCL13 levels in serum and synovial fluid with the radiographic severity of knee osteoarthritis. J. Investig. Med. 2015, 63, 545–547.
- Wei, F.; Moore, D.C.; Wei, L.; Li, Y.; Zhang, G.; Wei, X.; Lee, J.K.; Chen, Q. Correction: Attenuation of osteoarthritis via blockade of the SDF-1/CXCR4 signaling pathway. Arthritis Res. Ther. 2013, 15, 410.
More
Information
Subjects:
Orthopedics
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
10 Sep 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No