Osteoarthritis is a common cause of disability worldwide. Although commonly referred to as a disease of the joint cartilage, osteoarthritis affects all joint tissues equally. The pathogenesis of this degenerative process is not completely understood; however, a low-grade inflammation leading to an imbalance between anabolic and katabolic processes is a well-established factor. The complex network of cytokines regulating these processes and cell communication has a central role in the development and progression of osteoarthritis. Concentrations of both proinflammatory and anti-inflammatory cytokines were found to be altered depending on the osteoarthritis stage and activity.
- osteoarthritis
- cytokines
- chemokines
- pathogenesis
- inflammation
- biomarker
1. Introduction
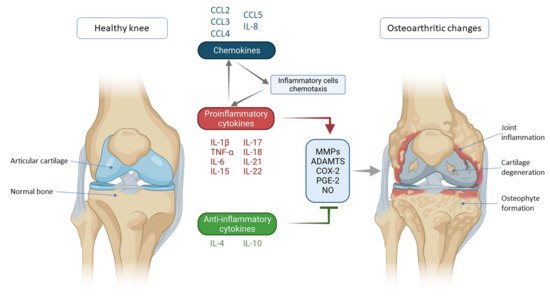
2. Cytokines and Chemokines Involved in Knee Osteoarthritis Pathogenesis
2.1. Proinflammatory Cytokines
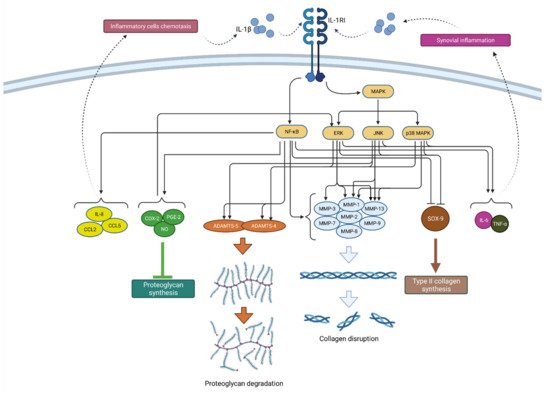
2.1.1. IL-1β
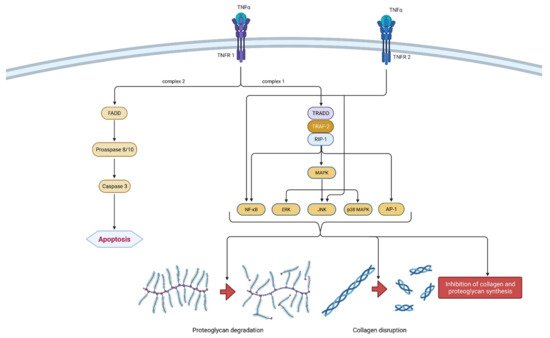
2.1.2. TNF-α
2.2. Anti-Inflammatory Cytokines
2.2.1. IL-4
2.2.2. IL-10
2.3. Chemokines
This entry is adapted from the peer-reviewed paper 10.3390/ijms22179208
References
- Neogi, T. The epidemiology and impact of pain in osteoarthritis. Osteoarthr. Cartil. 2013, 21, 1145–1153.
- Cui, A.; Li, H.; Wang, D.; Zhong, J.; Chen, Y.; Lu, H. Global, regional prevalence, incidence and risk factors of knee osteoarthritis in population-based studies. EClinicalMedicine 2020, 29–30, 100587.
- Primorac, D.; Molnar, V.; Rod, E.; Jeleč, Ž.; Čukelj, F.; Matišić, V.; Vrdoljak, T.; Hudetz, D.; Hajsok, H.; Borić, I. Knee Osteoarthritis: A Review of Pathogenesis and State-Of-The-Art Non-Operative Therapeutic Considerations. Genes 2020, 11, 854.
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.-P.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42.
- Scanzello, C.R. Chemokines and inflammation in osteoarthritis: Insights from patients and animal models. J. Orthop. Res. 2017, 35, 735–739.
- Fields, J.K.; Günther, S.; Sundberg, E.J. Structural Basis of IL-1 Family Cytokine Signaling. Front. Immunol. 2019, 10, 1412.
- Boraschi, D.; Italiani, P.; Weil, S.; Martin, M.U. The family of the interleukin-1 receptors. Immunol. Rev. 2018, 281, 197–232.
- Martel-Pelletier, J.; Mccollum, R.; Dibattista, J.; Faure, M.-P.; Chin, J.A.; Fournier, S.; Sarfati, M.; Pelletier, J.-P. The interleukin-1 receptor in normal and osteoarthritic human articular chondrocytes. Identification as the type I receptor and analysis of binding kinetics and biologic function. Arthritis Rheum. 1992, 35, 530–540.
- Attur, M.; Statnikov, A.; Samuels, J.; Li, Z.; Alekseyenko, A.V.; Greenberg, J.D.; Krasnokutsky, S.; Rybak, L.; Lu, Q.A.; Todd, J.; et al. Plasma levels of interleukin-1 receptor antagonist (IL1Ra) predict radiographic progression of symptomatic knee osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1915–1924.
- Wang, X.; Li, F.; Fan, C.; Wang, C.; Ruan, H. Effects and relationship of ERK1 and ERK2 in interleukin-1β-induced alterations in MMP3, MMP13, type II collagen and aggrecan expression in human chondrocytes. Int. J. Mol. Med. 2011, 27, 583–589.
- Hwang, S.-G.; Yu, S.-S.; Poo, H.; Chun, J.-S. c-Jun/Activator Protein-1 Mediates Interleukin-1β-induced Dedifferentiation but Not Cyclooxygenase-2 Expression in Articular Chondrocytes. J. Biol. Chem. 2005, 280, 29780–29787.
- Jenei-Lanzl, Z.; Meurer, A.; Zaucke, F. Interleukin-1β signaling in osteoarthritis—chondrocytes in focus. Cell. Signal. 2019, 53, 212–223.
- Hwang, H.; Kim, H. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2015, 16, 26035–26054.
- Wang, X.; Li, F.; Fan, C.; Wang, C.; Ruan, H. Analysis of isoform specific ERK signaling on the effects of interleukin-1β on COX-2 expression and PGE2 production in human chondrocytes. Biochem. Biophys. Res. Commun. 2010, 402, 23–29.
- Chow, Y.Y.; Chin, K.-Y. The Role of Inflammation in the Pathogenesis of Osteoarthritis. Mediat. Inflamm. 2020, 2020, 1–19.
- Choi, M.C.; Jo, J.; Park, J.; Kang, H.K.; Park, Y. NF-B Signaling Pathways in Osteoarthritic Cartilage Destruction. Cells 2019, 8, 734.
- Lepetsos, P.; Papavassiliou, K.A.; Papavassiliou, A.G. Redox and NF-κB signaling in osteoarthritis. Free Radic. Biol. Med. 2019, 132, 90–100.
- Chevalier, X.; Eymard, F.; Richette, P. Biologic agents in osteoarthritis: Hopes and disappointments. Nat. Rev. Rheumatol. 2013, 9, 400–410.
- Chevalier, X.; Goupille, P.; Beaulieu, A.D.; Burch, F.X.; Bensen, W.G.; Conrozier, T.; Loeuille, D.; Kivitz, A.J.; Silver, D.; Appleton, B.E. Intraarticular injection of anakinra in osteoarthritis of the knee: A multicenter, randomized, double-blind, placebo-controlled study. Arthritis Care Res. 2009, 61, 344–352.
- Chevalier, X.; Eymard, F. Anti-IL-1 for the treatment of OA: Dead or alive? Nat. Rev. Rheumatol. 2019, 15, 191–192.
- Theeuwes, W.F.; van den Bosch, M.H.J.; Thurlings, R.M.; Blom, A.B.; van Lent, P.L.E.M. The role of inflammation in mesenchymal stromal cell therapy in osteoarthritis, perspectives for post-traumatic osteoarthritis: A review. Rheumatology 2021, 60, 1042–1053.
- Baud, V.; Karin, M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001, 11, 372–377.
- Bodmer, J.L.; Schneider, P.; Tschopp, J. The molecular architecture of the TNF superfamily. Trends Biochem. Sci. 2002, 27, 19–26.
- Kriegler, M.; Perez, C.; DeFay, K.; Albert, I.; Lu, S.D. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: Ramifications for the complex physiology of TNF. Cell 1988, 53, 45–53.
- Zhou, T.; Mountz, J.D.; Kimberly, R.P. Immunobiology of tumor necrosis factor receptor superfamily. Immunol. Res. 2002, 26, 323–336.
- Appay, V.; Sauce, D. Immune activation and inflammation in HIV-1 infection: Causes and consequences. J. Pathol. 2008, 214, 231–241.
- Westacott, C.I.; Barakat, A.F.; Wood, L.; Perry, M.J.; Neison, P.; Bisbinas, I.; Armstrong, L.; Millar, A.B.; Elson, C.J. Tumor necrosis factor alpha can contribute to focal loss of cartilage in osteoarthritis. Osteoarthr. Cartil. 2000, 8, 213–221.
- Wojdasiewicz, P.; Poniatowski, Ł.A.; Szukiewicz, D. The Role of Inflammatory and Anti-Inflammatory Cytokines in the Pathogenesis of Osteoarthritis. Mediat. Inflamm. 2014, 2014, 1–19.
- Zelová, H.; Hošek, J. TNF-α signalling and inflammation: Interactions between old acquaintances. Inflamm. Res. 2013, 62, 641–651.
- Henderson, B.; Pettipher, E.R. Arthritogenic actions of recombinant IL-1 and tumour necrosis factor α in the rabbit: Evidence for synergistic interactions between cytokines in vivo. Clin. Exp. Immunol. 1989, 75, 306–310.
- Séguin, C.A.; Bernier, S.M. TNFα Suppresses Link Protein and Type II Collagen Expression in Chondrocytes: Role of MEK1/2 and NF-κB Signaling Pathways. J. Cell. Physiol. 2003, 197, 356–369.
- Xue, J.; Wang, J.; Liu, Q.; Luo, A. Tumor necrosis factor-α induces ADAMTS-4 expression in human osteoarthritis chondrocytes. Mol. Med. Rep. 2013, 8, 1755–1760.
- Oo, W.M.; Yu, S.P.-C.; Daniel, M.S.; Hunter, D.J. Disease-modifying drugs in osteoarthritis: Current understanding and future therapeutics. Expert Opin. Emerg. Drugs 2018, 23, 331–347.
- Brown, M.A.; Hural, J. Functions of IL-4 and control of its expression. Crit. Rev. Immunol. 2017, 37, 181–212.
- Bhattacharjee, A.; Shukla, M.; Yakubenko, V.P.; Mulya, A.; Kundu, S.; Cathcart, M.K. IL-4 and IL-13 employ discrete signaling pathways for target gene expression in alternatively activated monocytes/macrophages. Free Radic. Biol. Med. 2013, 54, 1–16.
- Forster, T.; Chapman, K.; Loughlin, J. Common variants within the interleukin 4 receptor α gene (IL4R) are associated with susceptibility to osteoarthritis. Hum. Genet. 2004, 114, 391–395.
- Schlaak, J.F.; Pfers, I.; Meyer Zum Büschenfelde, K.H.; Märker-Hermann, E. Different cytokine profiles in the synovial fluid of patients with osteoarthritis, rheumatoid arthritis and seronegative spondylarthropathies. Clin. Exp. Rheumatol. 1996, 14, 155–162.
- Wagner, S.; Fritz, P.; Einsele, H.; Sell, S.; Saal, J.G. Evaluation of synovial cytokine patterns in rheumatoid arthritis and osteoarthritis by quantitative reverse transcription polymerase chain reaction. Rheumatol. Int. 1997, 16, 191–196.
- Ishii, H.; Tanaka, H.; Katoh, K.; Nakamura, H.; Nagashima, M.; Yoshino, S. Characterization of infiltrating T cells and Th1/Th2-type cytokines in the synovium of patients with osteoarthritis. Osteoarthr. Cartil. 2002, 10, 277–281.
- Yeh, L.A.; Augustine, A.J.; Lee, P.; Riviere, L.R.; Sheldon, A. Interleukin-4, an inhibitor of cartilage breakdown in bovine articular cartilage explants. J. Rheumatol. 1995, 22, 1740–1746.
- Doi, H.; Nishida, K.; Yorimitsu, M.; Komiyama, T.; Kadota, Y.; Tetsunaga, T.; Yoshida, A.; Kubota, S.; Takigawa, M.; Ozaki, T. Interleukin-4 downregulates the cyclic tensile stress-induced matrix metalloproteinases-13 and cathepsin B expression by rat normal chondrocytes. Acta Med. Okayama 2008, 62, 119–126.
- Van Meegeren, M.E.R.; Roosendaal, G.; Jansen, N.W.D.; Wenting, M.J.G.; Van Wesel, A.C.W.; Van Roon, J.A.G.; Lafeber, F.P.J.G. IL-4 alone and in combination with IL-10 protects against blood-induced cartilage damage. Osteoarthr. Cartil. 2012, 20, 764–772.
- Schuerwegh, A.J.; Dombrecht, E.J.; Stevens, W.J.; Van Offel, J.F.; Bridts, C.H.; De Clerck, L.S. Influence of pro-inflammatory (IL-1α, IL-6, TNF-α, IFN-γ) and anti-inflammatory (IL-4) cytokines on chondrocyte function. Osteoarthr. Cartil. 2003, 11, 681–687.
- Schulze-Tanzil, G.; Zreiqat, H.; Sabat, R.; Kohl, B.; Halder, A.; Muller, R.; John, T. Interleukin-10 and Articular Cartilage: Experimental Therapeutical Approaches in Cartilage Disorders. Curr. Gene Ther. 2009, 9, 306–315.
- Mostafa, E.; Chollet-martin, S.; Oudghiri, M.; Laquay, N.; Jacob, M.; Michel, J.; Feldman, L.J. Effects of interleukin-10 on monocyte / endothelial cell adhesion and MMP-9/TIMP-1 secretion. Cardiovasc. Res. 2001, 49, 882–890.
- Wang, Y.S.; Wang, Y.H.; Zhao, G.Q.; Li, Y.B. Osteogenic potential of human calcitonin gene-related peptide alpha gene-modified bone marrow mesenchymal stem cells. Chin. Med. J. 2011, 124, 3976–3981.
- Behrendt, P.; Feldheim, M.; Preusse-Prange, A.; Weitkamp, J.T.; Haake, M.; Eglin, D.; Rolauffs, B.; Fay, J.; Seekamp, A.; Grodzinsky, A.J.; et al. Chondrogenic potential of IL-10 in mechanically injured cartilage and cellularized collagen ACI grafts. Osteoarthr. Cartil. 2018, 26, 264–275.
- Barker, T.; Rogers, V.E.; Henriksen, V.T.; Trawick, R.H.; Momberger, N.G.; Lynn Rasmussen, G. Circulating IL-10 is compromised in patients predisposed to developing and in patients with severe knee osteoarthritis. Sci. Rep. 2021, 11, 1–10.
- Helmark, I.C.; Mikkelsen, U.R.; Børglum, J.; Rothe, A.; Petersen, M.C.; Andersen, O.; Langberg, H.; Kjaer, M. Exercise increases interleukin-10 levels both intraarticularly and peri-synovially in patients with knee osteoarthritis: A randomized controlled trial. Arthritis Res. Ther. 2010, 12, R126.
- Fernandes, T.L.; Gomoll, A.H.; Lattermann, C.; Hernandez, A.J.; Bueno, D.F.; Amano, M.T. Macrophage: A Potential Target on Cartilage Regeneration. Front. Immunol. 2020, 11, 111.
- Zhang, J.; Rong, Y.; Luo, C.; Cui, W. Bone marrow mesenchymal stem cell-derived exosomes prevent osteoarthritis by regulating synovial macrophage polarization. Aging 2020, 12, 25138–25152.
- Watkins, L.R.; Chavez, R.A.; Landry, R.; Fry, M.; Green-Fulgham, S.M.; Coulson, J.D.; Collins, S.D.; Glover, D.K.; Rieger, J.; Forsayeth, J.R. Targeted interleukin-10 plasmid DNA therapy in the treatment of osteoarthritis: Toxicology and pain efficacy assessments. Brain. Behav. Immun. 2020, 90, 155–166.
- Borish, L.C.; Steinke, J.W. 2. Cytokines and chemokines. J. Allergy Clin. Immunol. 2003, 111, S460–S475.
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta- Mol. Cell Res. 2014, 1843, 2563–2582.
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interf. Cytokine Res. 2009, 29, 313–325.
- Monibi, F.; Roller, B.L.; Stoker, A.; Garner, B.; Bal, S.; Cook, J.L. Identification of Synovial Fluid Biomarkers for Knee Osteoarthritis and Correlation with Radiographic Assessment. J. Knee Surg. 2016, 29, 242–247.
- Watt, F.E.; Paterson, E.; Freidin, A.; Kenny, M.; Judge, A.; Saklatvala, J.; Williams, A.; Vincent, T.L. Acute Molecular Changes in Synovial Fluid Following Human Knee Injury: Association With Early Clinical Outcomes. Arthritis Rheumatol. 2016, 68, 2129–2140.
- Miller, R.E.; Tran, P.B.; Das, R.; Ghoreishi-Haack, N.; Ren, D.; Miller, R.J.; Malfait, A.M. CCR2 chemokine receptor signaling mediates pain in experimental osteoarthritis. Proc. Natl. Acad. Sci. USA 2012, 109, 20602–20607.
- Miller, R.E.; Belmadani, A.; Ishihara, S.; Tran, P.B.; Ren, D.; Miller, R.J.; Malfait, A.M. Damage-associated molecular patterns generated in osteoarthritis directly excite murine nociceptive neurons through toll-like receptor 4. Arthritis Rheumatol. 2015, 67, 2933–2943.
- Hulin-Curtis, S.L.; Bidwell, J.L.; Perry, M.J. Association between CCL2 haplotypes and knee osteoarthritis. Int. J. Immunogenet. 2013, 40, 280–283.
- Borzí, R.M.; Mazzetti, I.; Cattini, L.; Uguccioni, M.; Baggiolini, M.; Facchini, A. Human chondrocytes express functional chemokine receptors and release matrix-degrading enzymes in response to C-X-C and C-C chemokines. Arthritis Rheum. 2000, 43, 1734–1741.
- Zhao, X.Y.; Yang, Z.B.; Zhang, Z.J.; Zhang, Z.Q.; Kang, Y.; Huang, G.X.; Wang, S.W.; Huang, H.; Liao, W.M. CCL3 serves as a potential plasma biomarker in knee degeneration (osteoarthritis). Osteoarthr. Cartil. 2015, 23, 1405–1411.
- Beekhuizen, M.; Gierman, L.M.M.; van Spil, W.E.E.; Van Osch, G.J.V.M.J.V.M.; Huizinga, T.W.J.W.J.; Saris, D.B.F.B.F.; Creemers, L.B.B.; Zuurmond, A.-M.M. An explorative study comparing levels of soluble mediators in control and osteoarthritic synovial fluid. Osteoarthr. Cartil. 2013, 21, 918–922.
- Takebe, K.; Rai, M.F.; Schmidt, E.J.; Sandell, L.J. The chemokine receptor CCR5 plays a role in post-traumatic cartilage loss in mice, but does not affect synovium and bone. Osteoarthr. Cartil. 2015, 23, 454–461.
- Sandell, L.J.; Xing, X.; Franz, C.; Davies, S.; Chang, L.W.; Patra, D. Exuberant expression of chemokine genes by adult human articular chondrocytes in response to IL-1β. Osteoarthr. Cartil. 2008, 16, 1560–1571.
- Russo, R.C.; Garcia, C.C.; Teixeira, M.M.; Amaral, F.A. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev. Clin. Immunol. 2014, 10, 593–619.
- Merz, D.; Liu, R.; Johnson, K.; Terkeltaub, R. IL-8/CXCL8 and Growth-Related Oncogene α/CXCL1 Induce Chondrocyte Hypertrophic Differentiation. J. Immunol. 2003, 171, 4406–4415.
- Galliera, E.; Locati, M.; Mantovani, A.; Corsi, M.M. Chemokines and bone remodeling. Int. J. Immunopathol. Pharmacol. 2008, 21, 485–491.
- Sun, F.; Zhang, Y.; Li, Q. Therapeutic mechanisms of ibuprofen, prednisone and betamethasone in osteoarthritis. Mol. Med. Rep. 2017, 15, 981–987.
- Rasheed, Z.; Akhtar, N.; Haqqi, T.M. Advanced glycation end products induce the expression of interleukin-6 and interleukin-8 by receptor for advanced glycation end product-mediated activation of mitogen-activated protein kinases and nuclear factor-κB in human osteoarthritis chondrocytes. Rheumatology 2011, 50, 838–851.
- Takahashi, A.; de Andrés, M.C.; Hashimoto, K.; Itoi, E.; Oreffo, R.O.C. Epigenetic regulation of interleukin-8, an inflammatory chemokine, in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1946–1954.
- Frommer, K.W.; Hasseli, R.; Schäffler, A.; Lange, U.; Rehart, S.; Steinmeyer, J.; Rickert, M.; Sarter, K.; Zaiss, M.M.; Culmsee, C.; et al. Free Fatty Acids in Bone Pathophysiology of Rheumatic Diseases. Front. Immunol. 2019, 10, 2757.
- Yang, Y.; Gao, S.G.; Zhang, F.J.; Luo, W.; Xue, J.X.; Lei, G.H. Effects of osteopontin on the expression of IL-6 and IL-8 inflammatory factors in human knee osteoarthritis chondrocytes. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 3580–3586.
- Sakao, K.; Takahashi, K.A.; Arai, Y.; Saito, M.; Honjo, K.; Hiraoka, N.; Asada, H.; Shin-Ya, M.; Imanishi, J.; Mazda, O.; et al. Osteoblasts derived from osteophytes produce interleukin-6, interleukin-8, and matrix metalloproteinase-13 in osteoarthritis. J. Bone Miner. Metab. 2009, 27, 412–423.
- Remick, D.G.; DeForge, L.E.; Sullivan, J.F.; Showell, H.J. Profile of cytokines in synovial fluid specimens from patients with arthritis. Immunol. Invest. 1992, 21, 321–327.
- Lee, Y.A.; Choi, H.M.; Lee, S.H.; Yang, H.I.; Yoo, M.C.; Hong, S.J.; Kim, K.S. Synergy between adiponectin and interleukin-1β on the expression of interleukin-6, interleukin-8, and cyclooxygenase-2 in fibroblast-like synoviocytes. Exp. Mol. Med. 2012, 44, 440–447.
- Furuzawa-Carballeda, J.; Alcocer-Varela, J. Interleukin-8, interleukin- 10, intercellular adhesion molecule- 1 and vascular cell adhesion molecule-1 expression levels are higher in synovial tissue from patients with rheumatoid arthritis than in osteoarthritis. Scand. J. Immunol. 1999, 50, 215–222.
- Bertazzolo, N.; Punzi, L.; Stefani, M.P.; Cesaro, G.; Pianon, M.; Finco, B.; Todesco, S. Interrelationships between interleukin (IL)-1, IL-6 and IL-8 in synovial fluid of various arthropathies. Agents Actions 1994, 41, 90–92.
- Valcamonica, E.; Chighizola, C.B.; Comi, D.; De Lucia, O.; Pisoni, L.; Murgo, A.; Salvi, V.; Sozzani, S.; Meroni, P.L. Levels of chemerin and interleukin 8 in the synovial fluid of patients with inflammatory arthritides and osteoarthritis. Clin. Exp. Rheumatol. 2014, 32, 243–250.
- Kaneko, S.; Satoh, T.; Chiba, J.; Ju, C.; Inoue, K.; Kagawa, J. Interleukin-6 and interleukin-8 levels in serum and synovial fluid of patients with osteoarthritis. Cytokines Cell. Mol. Ther. 2000, 6, 71–79.
- Koh, S.M.M.; Chan, C.K.K.; Teo, S.H.H.; Singh, S.; Merican, A.; Ng, W.M.M.; Abbas, A.; Kamarul, T. Elevated plasma and synovial fluid interleukin-8 and interleukin-18 may be associated with the pathogenesis of knee osteoarthritis. Knee 2020, 27, 26–35.
- Allen, P.I.; Conzemius, M.G.; Evans, R.B.; Kiefer, K. Correlation between synovial fluid cytokine concentrations and limb function in normal dogs and in dogs with lameness from spontaneous osteoarthritis. Vet. Surg. 2019, 48, 770–779.
- Kleine, S.A.; Gogal, R.M.; George, C.; Thaliath, M.; Budsberg, S.C. Elevated Synovial Fluid Concentration of Monocyte Chemoattractant Protein-1 and Interleukin-8 in Dogs with Osteoarthritis of the Stifle. Vet. Comp. Orthop. Traumatol. 2020, 33, 147–150.
- García-Manrique, M.; Calvet, J.; Orellana, C.; Berenguer-Llergo, A.; Garcia-Cirera, S.; Llop, M.; Albiñana-Giménez, N.; Galisteo-Lencastre, C.; Gratacós, J. Synovial fluid but not plasma interleukin-8 is associated with clinical severity and inflammatory markers in knee osteoarthritis women with joint effusion. Sci. Rep. 2021, 11, 1–7.
- Ruan, G.; Xu, J.; Wang, K.; Zheng, S.; Wu, J.; Bian, F.; Chang, B.; Zhang, Y.; Meng, T.; Zhu, Z.; et al. Associations between serum IL-8 and knee symptoms, joint structures, and cartilage or bone biomarkers in patients with knee osteoarthritis. Clin. Rheumatol. 2019, 38, 3609–3617.
- Cecil, D.L.; Rose, D.M.; Terkeltaub, R.; Liu-Bryan, R. Role of interleukin-8 in PiT-1 expression and CXCR1-mediated inorganic phosphate uptake in chondrocytes. Arthritis Rheum. 2005, 52, 144–154.
- Marquez-Curtis, L.A.; Janowska-Wieczorek, A. Enhancing the migration ability of mesenchymal stromal cells by targeting the SDF-1/CXCR4 axis. Biomed Res. Int. 2013, 2013, 1–15.
- Shen, W.; Chen, J.; Zhu, T.; Chen, L.; Zhang, W.; Fang, Z.; Heng, B.C.; Yin, Z.; Chen, X.; Ji, J.; et al. Intra-Articular Injection of Human Meniscus Stem/Progenitor Cells Promotes Meniscus Regeneration and Ameliorates Osteoarthritis Through Stromal Cell-Derived Factor-1/CXCR4-Mediated Homing. Stem Cells Transl. Med. 2014, 3, 387–394.
- Gao, F.; Tian, J.; Pan, H.; Gao, J.; Yao, M. Association of CCL13 levels in serum and synovial fluid with the radiographic severity of knee osteoarthritis. J. Investig. Med. 2015, 63, 545–547.
- Wei, F.; Moore, D.C.; Wei, L.; Li, Y.; Zhang, G.; Wei, X.; Lee, J.K.; Chen, Q. Correction: Attenuation of osteoarthritis via blockade of the SDF-1/CXCR4 signaling pathway. Arthritis Res. Ther. 2013, 15, 410.