 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Charles G Mullighan | + 3153 word(s) | 3153 | 2021-08-31 05:40:23 | | | |
| 2 | Peter Tang | Meta information modification | 3153 | 2021-09-09 04:52:43 | | |
Video Upload Options
Acute lymphoblastic leukemia (ALL) is the most successful paradigm of how risk-adapted therapy and detailed understanding of the genetic alterations driving leukemogenesis and therapeutic response may dramatically improve treatment outcomes, with cure rates now exceeding 90% in children.
1. Introduction
2. B-Cell Precursor Acute Lymphoblastic Leukemia
2.1. Previously Established Subtypes with Recurring Chromosomal Abnormalities

2.2. Emerging B-ALL Subtypes Defined by Genome Sequencing Studies
2.3. Prognostic Implications
3. T-Cell Acute Lymphoblastic Leukemia (T-ALL)
3.1. Genomic Overview of T-ALL
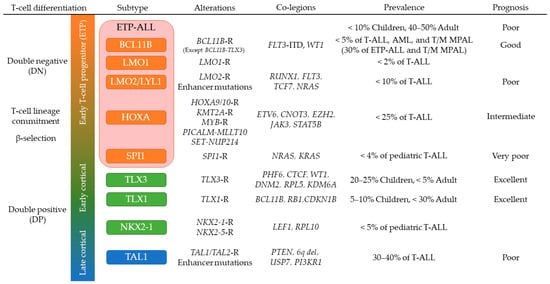
3.2. T-ALL in Early Stages of Cortical Thymocyte Maturation
3.3. TAL1-Driven T-ALL with Late Stages of Cortical Thymocyte Maturation
3.4. Early T-Cell Precursor (ETP) ALL and Mixed Phenotype Acute Leukemia
3.5. NOTCH1 Activating Mutations in T-ALL
4. Implications for Diagnosis
|
Platform |
Capability |
Cost |
Detectable Subtypes |
Difficult Subtypes |
|---|---|---|---|---|
|
WTS (RNAseq) |
Fusion chimeras Gene expression profiling Mutant allele expression Alternative splicing analysis (BCR/TCR rearrangements) (Sequence mutations) (Copy number analysis) |
Moderate |
B-ALL ETV6-RUNX1; KMT2A; TCF3-PBX1; BCR-ABL1; DUX4; MEF2D; ZNF384/362 NUTM1; HLF; BCL2/MYC; PAX5alt; ZEB2/CEBPE; -like subtypes |
B-ALL Aneuploidies |
|
T-ALL HOXA (KMT2A-R, PICALM-MLLT10, SET-NUP214); SPI1; NKX2-1; TAL1 (STIL-TAL1) |
T-ALL BCL11B; TLX1/3; LMO1/2; HOXA (others); TAL1 (others); T-other |
|||
|
WGS |
Sequence mutations Structural variants Copy number analysis (BCR/TCR rearrangements) (GWAS) |
High |
B-ALL Aneuploidies; ETV6-RUNX1; KMT2A; TCF3-PBX1; BCR-ABL1; DUX4; MEF2D; ZNF384/362; NUTM1; HLF; BCL2/MYC; PAX5 P80R; IKZF1 N159Y; ZEB2/CEBPE; Sequence and structural alterations in Ph-like ALL |
B-ALL -like subtypes; Part of PAX5alt |
|
T-ALL BCL11B; TLX1/3; LMO1/2; HOXA; SPI1; NKX2-1; TAL1 |
T-ALL T-other |
|||
|
WES |
Sequence mutations (coding) Structural variants (coding) Copy number analysis |
Moderate |
B-ALL (Aneuploidies) PAX5 P80R IKZF1 N159Y Sequence mutations in Ph-like ALL (e.g., JAK1/2/3, Ras) |
Most of other B-ALL and T-ALL subtypes |
|
Targeted sequencing (DNA and/or RNA) |
Fusion chimeras (targeted) Gene expression (targeted) Sequence mutations (targeted) Structural variants (targeted) (Copy number analysis) |
Low |
Targeted alterations |
Non-targeted alterations |
The parenthesis in “Capability” indicates analyses in development. Abbreviations: WTS: whole transcriptome sequencing; BCR: B-cell receptor; TCR: T-cell receptor; WGS: whole genome sequencing; GWAS: genome wide association study; WES: whole exome sequencing; -R: rearranged.
References
- Tran, T.H.; Hunger, S.P. The genomic landscape of pediatric acute lymphoblastic leukemia and precision medicine opportunities. Semin. Cancer Biol. 2020.
- Pui, C.-H. Precision medicine in acute lymphoblastic leukemia. Front. Med. 2020, 14, 689–700.
- Inaba, H.; Mullighan, C.G. Pediatric acute lymphoblastic leukemia. Haematologica 2020, 105, 2524.
- Stock, W.; Luger, S.M.; Advani, A.S.; Yin, J.; Harvey, R.C.; Mullighan, C.G.; Willman, C.L.; Fulton, N.; Laumann, K.M.; Malnassy, G.; et al. A pediatric regimen for older adolescents and young adults with acute lymphoblastic leukemia: Results of CALGB 10403. Blood 2019, 133, 1548–1559.
- Montefiori, L.E.; Bendig, S.; Gu, Z.; Chen, X.; Polonen, P.; Ma, X.; Murison, A.; Zeng, A.; Garcia-Prat, L.; Dickerson, K.; et al. Enhancer hijacking drives oncogenic BCL11B expression in lineage ambiguous stem cell leukemia. Cancer Discov. 2021.
- Iacobucci, I.; Mullighan, C.G. Genetic Basis of Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2017, 35, 975–983.
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019, 51, 296–307.
- Pui, C.-H.; Nichols, K.E.; Yang, J.J. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat. Rev. Clin. Oncol. 2018, 16, 227–240.
- Schwab, C.; Harrison, C.J. Advances in B-cell Precursor Acute Lymphoblastic Leukemia Genomics. HemaSphere 2018, 2, e53.
- Li, J.; Dai, Y.; Wu, L.; Zhang, M.; Ouyang, W.; Huang, J.; Chen, S. Emerging molecular subtypes and therapeutic targets in B-cell precursor acute lymphoblastic leukemia. Front. Med. 2021, 15, 347–371.
- Mullighan, C.G. How advanced are we in targeting novel subtypes of ALL? Best Pract. Res. Clin. Haematol. 2019, 32, 101095.
- Kimura, S.; Mullighan, C.G. Molecular markers in ALL: Clinical implications. Best Pract. Res. Clin. Haematol. 2020, 33, 101193.
- Li, J.-F.; Dai, Y.-T.; Lilljebjörn, H.; Shen, S.-H.; Cui, B.-W.; Bai, L.; Liu, Y.-F.; Qian, M.-X.; Kubota, Y.; Kiyoi, H.; et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1223 cases. Proc. Natl. Acad. Sci. USA 2018, 115, E11711–E11720.
- Roberts, K.G.; Morin, R.D.; Zhang, J.; Hirst, M.; Zhao, Y.; Su, X.; Chen, S.-C.; Payne-Turner, D.; Churchman, M.L.; Harvey, R.; et al. Genetic Alterations Activating Kinase and Cytokine Receptor Signaling in High-Risk Acute Lymphoblastic Leukemia. Cancer Cell 2012, 22, 153–166.
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.; Yang, Y.-L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2014, 371, 1005–1015.
- Klco, J.M.; Mullighan, C.G. Advances in germline predisposition to acute leukaemias and myeloid neoplasms. Nat. Rev. Cancer 2020, 21, 122–137.
- Perez-Andreu, V.; Roberts, K.G.; Harvey, R.; Yang, W.; Cheng, C.; Pei, D.; Xu, H.; Gastier-Foster, J.; Lim, J.Y.-S.; Chen, I.-M.; et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat. Genet. 2013, 45, 1494–1498.
- Ellinghaus, E.; Stanulla, M.; Richter, G.; Kronnie, G.T.; Cario, G.; Cazzaniga, G.; Horstmann, M.; Grümayer, R.P.; Cavé, H.; Trka, J.; et al. Identification of germline susceptibility loci in ETV6-RUNX1-rearranged childhood acute lymphoblastic leukemia. Leukemia 2011, 26, 902–909.
- Qian, M.; Zhao, X.; Devidas, M.; Yang, W.; Gocho, Y.; Smith, C.; Gastier-Foster, J.M.; Li, Y.; Xu, H.; Zhang, S.; et al. Genome-Wide Association Study of Susceptibility Loci for T-Cell Acute Lymphoblastic Leukemia in Children. J. Natl. Cancer Inst. 2019, 111, 1350–1357.
- Qian, M.; Xu, H.; Perez-Andreu, V.; Roberts, K.G.; Zhang, H.; Yang, W.; Zhang, S.; Zhao, X.; Smith, C.; Devidas, M.; et al. Novel susceptibility variants at the ERG locus for childhood acute lymphoblastic leukemia in Hispanics. Blood 2019, 133, 724–729.
- Paietta, E.; Roberts, K.G.; Wang, V.; Gu, Z.; Buck, G.; Pei, D.; Cheng, C.; Levine, R.L.; Abdel-Wahab, O.; Cheng, Z.; et al. Molecular Classification Improves Risk Assessment in Adult BCR-ABL1-negative B-ALL. Blood 2021.
- Jeha, S.; Choi, J.; Roberts, K.G.; Pei, D.; Coustan-Smith, E.; Inaba, H.; Rubnitz, J.E.; Ribeiro, R.C.; Gruber, T.A.; Raimondi, S.C.; et al. Clinical Significance of Novel Subtypes of Acute Lymphoblastic Leukemia in the Context of Minimal Residual Disease–Directed Therapy. Blood Cancer Discov. 2021, 2, 326–337.
- Sirvent, N.; Trassard, M.; Ebran, N.; Attias, R.; Pedeutour, F. Fusion of EWSR1 with the DUX4 facioscapulohumeral muscular dystrophy region resulting from t(4;22)(q35;q12) in a case of embryonal rhabdomyosarcoma. Cancer Genet. Cytogenet. 2009, 195, 12–18.
- Aifantis, I.; Raetz, E.A.; Buonamici, S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat. Rev. Immunol. 2008, 8, 380–390.
- Yui, M.; Rothenberg, E. Developmental gene networks: A triathlon on the course to T cell identity. Nat. Rev. Immunol. 2014, 14, 529–545.
- Teachey, D.T.; Pui, C.-H. Comparative features and outcomes between paediatric T-cell and B-cell acute lymphoblastic leukaemia. Lancet Oncol. 2019, 20, e142–e154.
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218.
- Seki, M.; Kimura, S.; Isobe, T.; Yoshida, K.; Ueno, H.; Nakajima-Takagi, Y.; Wang, C.; Lin, L.; Kon, A.; Suzuki, H.; et al. Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1274–1281.
- Mansour, M.; Abraham, B.; Anders, L.; Berezovskaya, A.; Gutierrez, A.; Durbin, A.; Etchin, J.; Lawton, L.; Sallan, S.E.; Silverman, L.B.; et al. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 2014, 346, 1373–1377.
- Hu, S.; Qian, M.; Zhang, H.; Guo, Y.; Yang, J.; Zhao, X.; He, H.; Lu, J.; Pan, J.; Chang, M.; et al. Whole-genome noncoding sequence analysis in T-cell acute lymphoblastic leukemia identifies oncogene enhancer mutations. Blood 2017, 129, 3264–3268.
- Mansour, M.R.; Duke, V.; Foroni, L.; Patel, B.; Allen, C.; Ancliff, P.J.; Gale, R.E.; Linch, D.C. Notch-1 Mutations Are Secondary Events in Some Patients with T-Cell Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2007, 13, 6964–6969.
- Gianni, F.; Belver, L.; Ferrando, A. The Genetics and Mechanisms of T-Cell Acute Lymphoblastic Leukemia. Cold Spring Harb. Perspect. Med. 2019, 10, a035246.
- Belver, L.; Ferrando, L.B.A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2016, 16, 494–507.
- Kimura, S.; Seki, M.; Kawai, T.; Goto, H.; Yoshida, K.; Isobe, T.; Sekiguchi, M.; Watanabe, K.; Kubota, Y.; Nannya, Y.; et al. DNA methylation-based classification reveals difference between pediatric T-cell acute lymphoblastic leukemia and normal thymocytes. Leukemia 2019, 34, 1163–1168.
- Roels, J.; Thénoz, M.; Szarzyńska, B.; Landfors, M.; De Coninck, S.; Demoen, L.; Provez, L.; Kuchmiy, A.; Strubbe, S.; Reunes, L.; et al. Aging of Preleukemic Thymocytes Drives CpG Island Hypermethylation in T-cell Acute Lymphoblastic Leukemia. Blood Cancer Discov. 2020, 1, 274–289.
- Ferrando, A.A.; Neuberg, D.S.; Staunton, J.; Loh, M.L.; Huard, C.; Raimondi, S.C.; Behm, F.G.; Pui, C.-H.; Downing, J.R.; Gilliland, D.; et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002, 1, 75–87.
- Soulier, J.; Clappier, E.; Cayuela, J.-M.; Regnault, A.; García-Peydró, M.; Dombret, H.; Baruchel, A.; Toribio, M.L.; Sigaux, F. HOXA genes are included in genetic and biologic networks defining human acute T-cell leukemia (T-ALL). Blood 2005, 106, 274–286.
- Dadi, S.; Le Noir, S.; Bornet, D.P.; Lhermitte, L.; Zacarias-Cabeza, J.; Bergeron, J.; Villarèse, P.; Vachez, E.; Dik, W.A.; Millien, C.; et al. TLX Homeodomain Oncogenes Mediate T Cell Maturation Arrest in T-ALL via Interaction with ETS1 and Suppression of TCRα Gene Expression. Cancer Cell 2012, 21, 563–576.
- Di Giacomo, D.; La Starza, R.; Gorello, P.; Pellanera, F.; Atak, Z.K.; De Keersmaecker, K.; Pierini, V.; Harrison, C.J.; Arniani, S.; Moretti, M.; et al. 14q32 rearrangements deregulating BCL11B mark a distinct subgroup of T and myeloid immature acute leukemia. Blood 2021.
- Inaba, H.; Azzato, E.M.; Mullighan, C.G. Integration of Next-Generation Sequencing to Treat Acute Lymphoblastic Leukemia with Targetable Lesions: The St. Jude Children’s Research Hospital Approach. Front. Pediatr. 2017, 5, 258.
- Gocho, Y.; Liu, J.; Hu, J.; Yang, W.; Dharia, N.V.; Zhang, J.; Shi, H.; Du, G.; John, A.; Lin, T.-N.; et al. Network-based systems pharmacology reveals heterogeneity in LCK and BCL2 signaling and therapeutic sensitivity of T-cell acute lymphoblastic leukemia. Nat. Rev. Cancer 2021, 2, 1–16.
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.-L.; Bak, R.; Li, C.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 2016, 351, 1454–1458.
- Park, S.T.; Sun, X.-H. The Tal1 Oncoprotein Inhibits E47-mediated Transcription. J. Biol. Chem. 1998, 273, 7030–7037.
- Sanda, T.; Lawton, L.N.; Barrasa, M.I.; Fan, Z.P.; Kohlhammer, H.; Gutierrez, A.; Ma, W.; Tatarek, J.; Ahn, Y.; Kelliher, M.A.; et al. Core Transcriptional Regulatory Circuit Controlled by the TAL1 Complex in Human T Cell Acute Lymphoblastic Leukemia. Cancer Cell 2012, 22, 209–221.
- Nottingham, W.T.; Jarratt, A.; Burgess, M.; Speck, C.L.; Cheng, J.F.; Prabhakar, S.; Rubin, E.M.; Li, P.S.; Sloane-Stanley, J.; Kong, A.S.J.; et al. Runx1-mediated hematopoietic stem-cell emergence is controlled by a Gata/Ets/SCL-regulated enhancer. Blood 2007, 110, 4188–4197.
- Choi, A.; Illendula, A.; Pulikkan, J.A.; Roderick, J.E.; Tesell, J.; Yu, J.; Hermance, N.; Zhu, L.J.; Castilla, L.H.; Bushweller, J.H.; et al. RUNX1 is required for oncogenic Myb and Myc enhancer activity in T-cell acute lymphoblastic leukemia. Blood 2017, 130, 1722–1733.
- O’Neil, J.; Shank, J.; Cusson, N.; Murre, C.; Kelliher, M. TAL1/SCL induces leukemia by inhibiting the transcriptional activity of E47/HEB. Cancer Cell 2004, 5, 587–596.
- Draheim, K.M.; Hermance, N.; Yang, Y.; Arous, E.; Calvo, J.; Kelliher, M.A. A DNA-binding mutant of TAL1 cooperates with LMO2 to cause T cell leukemia in mice. Oncogene 2010, 30, 1252–1260.
- Ferrando, A.A.; Look, A.T. Gene expression profiling in T-cell acute lymphoblastic leukemia. Semin. Hematol. 2003, 40, 274–280.
- Leong, W.Z.; Tan, S.H.; Ngoc, P.C.T.; Amanda, S.; Yam, A.W.Y.; Liau, W.-S.; Gong, Z.; Lawton, L.N.; Tenen, D.G.; Sanda, T. ARID5B as a critical downstream target of the TAL1 complex that activates the oncogenic transcriptional program and promotes T-cell leukemogenesis. Genes Dev. 2017, 31, 2343–2360.
- Tan, S.H.; Leong, W.Z.; Ngoc, P.C.T.; Tan, T.K.; Bertulfo, F.C.; Lim, M.C.; An, O.; Li, Z.; Yeoh, A.E.J.; Fullwood, M.J.; et al. The enhancer RNA ARIEL activates the oncogenic transcriptional program in T-cell acute lymphoblastic leukemia. Blood 2019, 134, 239–251.
- Piovan, E.; Yu, J.; Tosello, V.; Herranz, D.; Ambesi-Impiombato, A.; Da Silva, A.C.; Sanchez-Martin, M.; Perez-Garcia, A.; Rigo, I.; Castillo, M.; et al. Direct Reversal of Glucocorticoid Resistance by AKT Inhibition in Acute Lymphoblastic Leukemia. Cancer Cell 2013, 24, 766–776.
- Jena, N.; Sheng, J.; Hu, J.K.; Li, W.; Zhou, W.; Lee, G.; Tsichlis, N.; Pathak, A.; Brown, N.; Deshpande, A.; et al. CDK6-mediated repression of CD25 is required for induction and maintenance of Notch1-induced T-cell acute lymphoblastic leukemia. Leukemia 2015, 30, 1033–1043.
- Coustan-Smith, E.; Mullighan, C.; Onciu, M.; Behm, F.G.; Raimondi, S.C.; Pei, D.; Cheng, C.; Su, X.; Rubnitz, J.; Basso, G.; et al. Early T-cell precursor leukaemia: A subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009, 10, 147–156.
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163.
- Alexander, T.B.; Gu, Z.; Iacobucci, I.; Dickerson, K.; Choi, J.K.; Xu, B.; Payne-Turner, D.; Yoshihara, H.; Loh, M.L.; Horan, J.; et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 2018, 562, 373–379.
- Maude, S.L.; Dolai, S.; Delgado-Martin, C.; Vincent, T.; Robbins, A.; Selvanathan, A.; Ryan, T.; Hall, J.; Wood, A.C.; Tasian, S.K.; et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood 2015, 125, 1759–1767.
- McCarter, A.C.; Wang, Q.; Chiang, M. Notch in Leukemia. Adv. Exp. Med. Biol. 2018, 1066, 355–394.
- Weng, A.; Ferrando, A.A.; Lee, W.; Iv, J.P.M.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating Mutations of NOTCH1 in Human T Cell Acute Lymphoblastic Leukemia. Science 2004, 306, 269–271.
- Sulis, M.L.; Williams, O.; Palomero, T.; Tosello, V.; Pallikuppam, S.; Real, P.J.; Barnes, K.; Zuurbier, L.; Meijerink, J.P.; Ferrando, A.A. NOTCH1 extracellular juxtamembrane expansion mutations in T-ALL. Blood 2008, 112, 733–740.
- Haydu, J.E.; De Keersmaecker, K.; Duff, M.K.; Paietta, E.; Racevskis, J.; Wiernik, P.H.; Rowe, J.M.; Ferrando, A. An activating intragenic deletion in NOTCH1 in human T-ALL. Blood 2012, 119, 5211–5214.
- Thompson, B.J.; Buonamici, S.; Sulis, M.L.; Palomero, T.; Vilimas, T.; Basso, G.; Ferrando, A.; Aifantis, I. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J. Exp. Med. 2007, 204, 1825–1835.
- O’Neil, J.; Grim, J.; Strack, P.; Rao, S.; Tibbitts, D.; Winter, C.; Hardwick, J.; Welcker, M.; Meijerink, J.; Pieters, R.; et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to γ-secretase inhibitors. J. Exp. Med. 2007, 204, 1813–1824.
- Suarez-Puente, X.; Beà, S.; Valdés-Mas, R.; Villamor, N.; Gutiérrez-Abril, J.; Martin-Subero, J.I.; Munar, M.; Rubio-Perez, C.; Jares, P.; Aymerich, M.; et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015, 526, 519–524.
- Chiang, M.; Xu, L.; Shestova, O.; Histen, G.; L’Heureux, S.; Romany, C.; Childs, M.E.; Gimotty, P.A.; Aster, J.C.; Pear, W.S. Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras–initiated leukemia. J. Clin. Investig. 2008, 118, 3181–3194.
- Wendorff, A.A.; Quinn, S.A.; Rashkovan, M.; Madubata, C.J.; Ambesi-Impiombato, A.; Litzow, M.R.; Tallman, M.S.; Paietta, E.; Paganin, M.; Basso, G.; et al. Phf6 Loss Enhances HSC Self-Renewal Driving Tumor Initiation and Leukemia Stem Cell Activity in T-ALL. Cancer Discov. 2018, 9, 436–451.
- Herranz, D.; Ambesi-Impiombato, A.; Palomero, T.; Schnell, S.A.; Belver, L.; Wendorff, A.A.; Xu, L.; Castillo-Martin, M.; Llobet-Navás, D.; Cordon-Cardo, C.; et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 2014, 20, 1130–1137.
- Albertí-Servera, L.; Demeyer, S.; Govaerts, I.; Swings, T.; De Bie, J.; Gielen, O.; Brociner, M.; Michaux, L.; Maertens, J.; Uyttebroeck, A.; et al. Single-cell DNA amplicon sequencing reveals clonal heterogeneity and evolution in T-cell acute lymphoblastic leukemia. Blood 2021, 137, 801–811.
- De Bie, J.; Demeyer, S.; Alberti-Servera, L.; Geerdens, E.; Segers, H.; Broux, M.; De Keersmaecker, K.; Michaux, L.; Vandenberghe, P.; Voet, T.; et al. Single-cell sequencing reveals the origin and the order of mutation acquisition in T-cell acute lymphoblastic leukemia. Leukemia 2018, 32, 1358–1369.
- Yashiro-Ohtani, Y.; Wang, H.; Zang, C.; Arnett, K.L.; Bailis, W.; Ho, Y.; Knoechel, B.; Lanauze, C.; Louis, L.; Forsyth, K.; et al. Long-range enhancer activity determinesMycsensitivity to Notch inhibitors in T cell leukemia. Proc. Natl. Acad. Sci. USA 2014, 111, E4946–E4953.
- Sanchez-Martin, M.; Ferrando, A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 2017, 129, 1124–1133.
- Reizis, B. Direct induction of T lymphocyte-specific gene expression by the mammalian Notch signaling pathway. Genes Dev. 2002, 16, 295–300.
- Zheng, R.; Li, M.; Wang, S.; Liu, Y. Advances of target therapy on NOTCH1 signaling pathway in T-cell acute lymphoblastic leukemia. Exp. Hematol. Oncol. 2020, 9, 31.
- Papayannidis, C.; De Angelo, D.J.; Stock, W.; Huang, B.; Shaik, M.N.; Cesari, R.; Zheng, X.; Reynolds, J.M.; English, P.A.; Ozeck, M.; et al. A Phase 1 study of the novel gamma-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J. 2015, 5, e350.
- Gavai, A.V.; Quesnelle, C.A.; Norris, D.J.; Han, W.-C.; Gill, P.S.; Shan, W.; Balog, A.; Chen, K.; Tebben, A.J.; Rampulla, R.; et al. Discovery of Clinical Candidate BMS-906024: A Potent Pan-Notch Inhibitor for the Treatment of Leukemia and Solid Tumors. ACS Med. Chem. Lett. 2015, 6, 523–527.
- Sanchez-Martin, M.; Ambesi-Impiombato, A.; Qin, Y.; Herranz, D.; Bansal, M.; Girardi, T.; Paietta, E.; Tallman, M.S.; Rowe, J.M.; De Keersmaecker, K.; et al. Synergistic antileukemic therapies inNOTCH1-induced T-ALL. Proc. Natl. Acad. Sci. USA 2017, 114, 2006–2011.
- Wei, P.; Walls, M.; Qiu, M.; Ding, R.; Denlinger, R.H.; Wong, A.; Tsaparikos, K.; Jani, J.P.; Hosea, N.A.; Sands, M.; et al. Evaluation of Selective γ-Secretase Inhibitor PF-03084014 for Its Antitumor Efficacy and Gastrointestinal Safety to Guide Optimal Clinical Trial Design. Mol. Cancer Ther. 2010, 9, 1618–1628.
- Real, P.; Tosello, V.; Palomero, T.; Castillo, M.; Hernando, E.; De Stanchina, E.; Sulis, M.L.; Barnes, K.; Sawai, C.; Homminga, I.; et al. γ-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat. Med. 2008, 15, 50–58.
- Cullion, K.; Draheim, K.M.; Hermance, N.; Tammam, J.; Sharma, V.M.; Ware, C.; Nikov, G.; Krishnamoorthy, V.; Majumder, P.K.; Kelliher, M.A. Targeting the Notch1 and mTOR pathways in a mouse T-ALL model. Blood 2009, 113, 6172–6181.
- Franciosa, G.; Smits, J.G.A.; Minuzzo, S.; Martinez-Val, A.; Indraccolo, S.; Olsen, J.V. Proteomics of resistance to Notch1 inhibition in acute lymphoblastic leukemia reveals targetable kinase signatures. Nat. Commun. 2021, 12, 2507.
- Trinquand, A.; Tanguy-Schmidt, A.; Ben Abdelali, R.; Lambert, J.; Beldjord, K.; Lengliné, E.; De Gunzburg, N.; Bornet, D.P.; Lhermitte, L.; Mossafa, H.; et al. Toward a NOTCH1/FBXW7/RAS/PTEN–Based Oncogenetic Risk Classification of Adult T-Cell Acute Lymphoblastic Leukemia: A Group for Research in Adult Acute Lymphoblastic Leukemia Study. J. Clin. Oncol. 2013, 31, 4333–4342.
- Newman, S.; Nakitandwe, J.; Kesserwan, C.A.; Azzato, E.M.; Wheeler, D.A.; Rusch, M.; Shurtleff, S.; Hedges, D.J.; Hamilton, K.V.; Foy, S.G.; et al. Genomes for Kids: The scope of pathogenic mutations in pediatric cancer revealed by comprehensive DNA and RNA sequencing. Cancer Discov. 2021.
- Jobanputra, V.; Wrzeszczynski, K.O.; Buttner, R.; Caldas, C.; Cuppen, E.; Grimmond, S.; Haferlach, T.; Mullighan, C.; Schuh, A.; Elemento, O. Clinical interpretation of whole-genome and whole-transcriptome sequencing for precision oncology. Semin. Cancer Biol. 2021.
- Rosenquist, R.; Cuppen, E.; Buettner, R.; Caldas, C.; Dreau, H.; Elemento, O.; Frederix, G.; Grimmond, S.; Haferlach, T.; Jobanputra, V.; et al. Clinical utility of whole-genome sequencing in precision oncology. Semin. Cancer Biol. 2021.
- Meggendorfer, M.; Jobanputra, V.; Wrzeszczynski, K.O.; Roepman, P.; de Bruijn, E.; Cuppen, E.; Buttner, R.; Caldas, C.; Grimmond, S.; Mullighan, C.G.; et al. Analytical demands to use whole-genome sequencing in precision oncology. Semin. Cancer Biol. 2021.


