 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yugeesh R Lankadeva | + 2662 word(s) | 2662 | 2021-09-08 05:06:39 | | | |
| 2 | Lindsay Dong | Meta information modification | 2662 | 2021-09-09 03:56:02 | | | | |
| 3 | Camila Xu | Meta information modification | 2662 | 2021-10-11 03:43:01 | | |
Video Upload Options
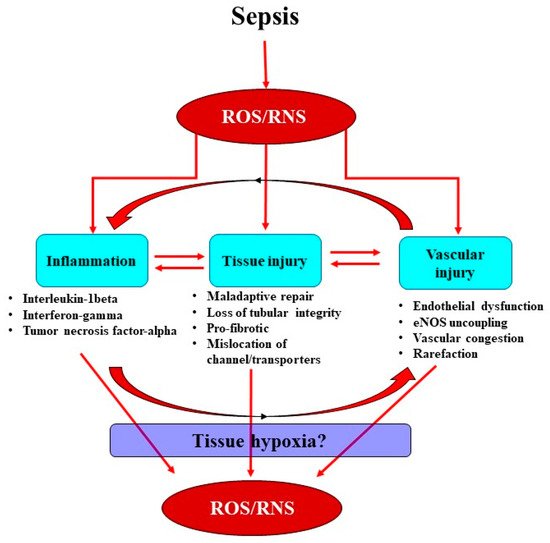
Sepsis is the leading cause of acute kidney injury (AKI) and leads to increased morbidity and mortality in intensive care units. Current treatments for septic AKI are largely supportive and are not targeted towards its pathophysiology. Sepsis is commonly characterized by systemic inflammation and increased production of reactive oxygen species (ROS), particularly superoxide. Concomitantly released nitric oxide (NO) then reacts with superoxide, leading to the formation of reactive nitrogen species (RNS), predominantly peroxynitrite. Sepsis-induced ROS and RNS can reduce the bioavailability of NO, mediating renal microcirculatory abnormalities, localized tissue hypoxia and mitochondrial dysfunction, thereby initiating a propagating cycle of cellular injury culminating in AKI.
1. Introduction
2. Interactions between the Septic Inflammatory Cascade and Oxidative Stress
3. Oxidative Stress Exacerbates Microcirculatory Abnormalities and Vascular Rarefaction
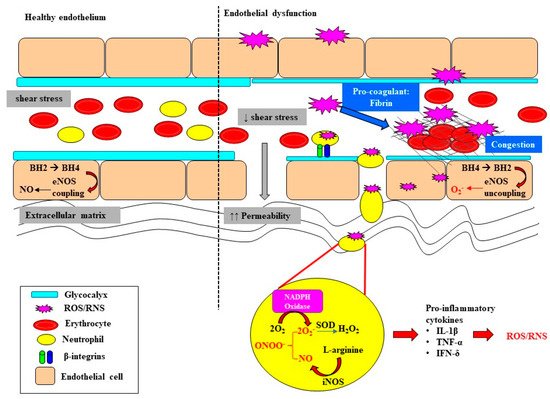
Endothelial dysfunction and microvascular rarefaction have been described as common pathophysiological features of AKI and are postulated to be critical factors mediating progression to CKD following recovery from AKI [30][31][32]. The NO system is an important regulator of vascular tone within the renal microcirculation, but it can be deleteriously affected in sepsis (Figure 2). In the healthy state, the biosynthesis of NO by vascular endothelial cells is dependent on the coupling state of endothelial nitric oxide synthase (eNOS) and the bioavailability of the co-factor tetrahydrobiopterin (BH4) [33]. When endogenous levels of the co-factor BH4 are sufficient, L-arginine is coupled with the reduction of oxygen, leading to the production of the potent vasodilator NO [33][34]. However, when BH4 levels are low, eNOS is uncoupled and superoxide is produced instead. Furthermore, BH4 is highly susceptible to oxidization to BH2 when levels of superoxide are high, further depleting the pool of the rate limiting co-factor BH4 [35][36]. Uncoupling of eNOS is reported to contribute to the pathophysiology of a myriad of kidney diseases arising from diabetes, hypertension and ischemia-reperfusion injury [37][38]. Intravenous supplementation with BH4 in an ovine model of sepsis improved microvascular dysfunction via increasing the number of perfused vessels, the proportion of perfused small vessels and the microvascular index within the sublingual circulation [39]. In an ovine model of severe septic AKI induced by intravenous infusion of live Escherichia Coli for 48 h, eNOS gene expression was selectively down regulated in the renal medulla, but not the renal cortex [11]. However, whether an uncoupling of eNOS contributes to the early onset of microcirculatory abnormalities reported within the renal medulla in ovine septic AKI [13] warrants further investigation.
In sepsis, excessive superoxide generation and accumulation, in tandem with inflammation, also results in direct structural damage to the vasculature, resulting in vascular leakage and tissue edema (Figure 2) [40].
The damaged endothelium also attracts leukocytes to the site of injury, as part of the innate immune response facilitated by the exposed intercellular and vascular cell adhesion molecules. This homing of pro-inflammatory cells, in conjunction with compromised gap junctions, leads to extravasation of the pro-inflammatory cells from the endothelium into the surrounding tissue, contributing to persistent inflammation [41]. Notably, inflammatory cells can generate ROS themselves and so reduce NO bioavailability [41], thereby contributing to the extensive pool of superoxide, essentially setting up a vicious cycle of oxidative stress, inflammation and vascular injury [19] (Figure 1). Sepsis-induced microvascular injury can also release microparticles into the systemic circulation.
4. Renal Medullary Tissue Hypoxia: A Critical Event in Acute Kidney Injury?
Renal medullary hypoxia is emerging as a common pathophysiological feature of AKI arising from sepsis [13][42], cardiopulmonary bypass [43][44] and radiocontrast-induced nephropathy [45]. Furthermore, renal medullary hypoxia has been implicated as an important driver in the transition and/or propensity for progression from AKI to CKD [46][47]. The relatively high metabolic requirements of the tubular elements in the renal medulla, coupled with the topography of vascular and tubular architecture within the medulla, result in a steep oxygen gradient between the capillaries (vasa recta) and both the thick and thin ascending limbs of the loop of Henle and the collecting ducts [48]. There is also the potential for diffusive oxygen shunting in the renal medullary microcirculation (from descending to ascending vasa recta), which could further compromise renal medullary oxygen delivery [49]. In healthy sheep, graded occlusion of the renal artery and thus progressive reductions in renal blood flow resulted in proportionally greater degrees of renal medullary ischemia and hypoxia compared with a renal cortex indicative of an intrinsic deficit in the autoregulatory capacity of the renal medullary microcirculation [50]. Accordingly, under pathophysiological settings such as sepsis, renal medullary microcirculatory perturbations leading to even modest reductions in medullary oxygen delivery or increases in oxygen consumption can have adverse consequences for medullary tissue oxygenation.
Renal medullary hypoxia can be a major driver of a cascade of events leading to cellular injury, vascular injury and tubular dysfunction [51]. Acute renal insults, including endotoxemia, can both increase renal tissue oxygen consumption and reduce tissue oxygen delivery. For example, these changes can result in tubular injury and obstruction, and mislocalization of Na/K-ATPase and transport proteins within renal tubular epithelial cells, thereby reducing the efficiency of oxygen utilization for sodium reabsorption [52].
5. Sepsis-Induced Mitochondrial Dysfunction Activates Production of ROS
Mitochondrial dysfunction is proposed to be both a cause and consequence of renal hypoxia in the pathogenesis of septic AKI [53][54]. Mitochondria are the main consumers of oxygen within the kidneys. Thus, the production of physiological levels of mitochondrial ROS in the mitochondrial matrix is important because ROS serve as signals and regulators for a myriad of biological processes.
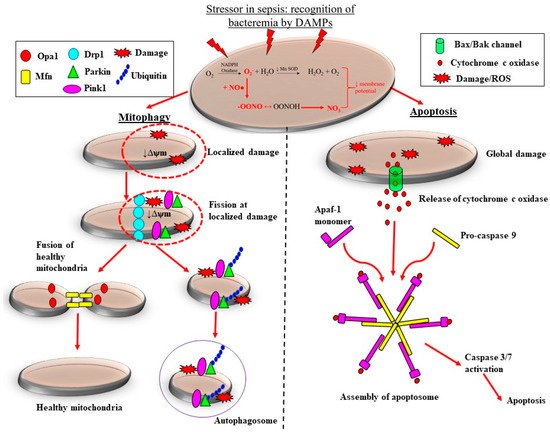
However, as cells experience prolonged periods of hypoxia, there is a change in metabolism and poor utilization of the available oxygen for ATP production in the mitochondrial electron transport chain, resulting in increased leakage of electrons and elevated production of free radicals/ROS [55][56]. Mitochondria use oxygen as the final acceptor of the respiratory chain, but its incomplete reduction can also produce ROS, especially superoxide [57]. Complex III of the electron transport chain is the inherent oxygen sensor during acute hypoxia, and it regulates the production of superoxide inversely with oxygen availability [58]. The transition of complex I from the active to “de-active” form was also reported to have the capacity to produce ROS outbursts during acute hypoxia [59]. Patients with septic AKI have elevated levels of receptor-interacting protein kinase-3 (RIPK3) in urine and plasma [60]. RIPK3 promotes oxidative stress and mitochondrial dysfunction in kidney tubular epithelial cells by increasing the expression and mitochondrial translocation of NADPH oxidase 4 and inhibition of mitochondrial complexes I and III [60][61][62]. It is therefore not surprising that mitochondrial injury has been commonly related to multi-organ dysfunction in patients with sepsis [6][63].
There are pre-clinical and clinical studies demonstrating that the adaptive processes of mitochondrial fission are downregulated in sepsis, which likely contributes to the loss of mitochondrial mass, thereby propagating ROS-induced damage during septic AKI. Sepsis is associated with considerable morphological changes in mitochondria. These changes include reduced numbers of cristae due to swelling of the inter-cristae space and the mitochondrial matrix, and vacuolation within the mitochondria space [60][64]. Depending on the severity of mitochondrial damage, the removal of mitochondria can be carried out by two pathways: mitophagy and apoptosis (Figure 3).
References
- Bagshaw, S.M.; George, C.; Dinu, I.; Bellomo, R. A multi-centre evaluation of the RIFLE criteria for early acute kidney injury in critically ill patients. Nephrol. Dial. Transplant. 2007, 23, 1203–1210.
- Bellomo, R.; Kellum, J.A.; Ronco, C.; Wald, R.; Martensson, J.; Maiden, M.; Bagshaw, S.M.; Glassford, N.J.; Lankadeva, Y.R.; Vaara, S.T.; et al. Acute kidney injury in sepsis. Intensive Care Med. 2017, 43, 816–828.
- Odutayo, A.; Wong, C.X.; Farkouh, M.; Altman, D.G.; Hopewell, S.; Emdin, C.A.; Hunn, B.H. AKI and long-rerm risk for cardiovascular events and mortality. J. Am. Soc. Nephrol. 2017, 28, 377–387.
- Rhodes, A.; Evans, L.E.; Alhazzani, W.; Levy, M.M.; Antonelli, M.; Ferrer, R.; Kumar, A.; Sevransky, J.E.; Sprung, C.L.; Nunnally, M.E.; et al. Surviving sepsis campaign: International guidelines for management of sepsis and septic shock. Intensive Care Med. 2017, 43, 304–377.
- Schrier, R.W.; Wang, W. Acute Renal Failure and Sepsis. N. Engl. J. Med. 2004, 351, 159–169.
- Takasu, O.; Gaut, J.P.; Watanabe, E.; To, K.; Fagley, R.E.; Sato, B.; Jarman, S.; Efimov, I.R.; Janks, D.L.; Srivastava, A.; et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am. J. Respir. Crit. Care Med. 2013, 187, 509–517.
- Lerolle, N.; Nochy, D.; Guérot, E.; Bruneval, P.; Fagon, J.-Y.; Diehl, J.-L.; Hill, G. Histopathology of septic shock induced acute kidney injury: Apoptosis and leukocytic infiltration. Intensive Care Med. 2009, 36, 471–478.
- Corrêa, T.D.; Jeger, V.; Pereira, A.J.; Takala, J.; Djafarzadeh, S.; Jakob, S.M. Angiotensin II in septic shock: Effects on tissue perfusion, organ function, and mitochondrial respiration in a porcine model of fecal peritonitis. Crit. Care Med. 2014, 42, e550–e559.
- Maiden, M.J.; Otto, S.; Brealey, J.K.; Finnis, M.E.; Chapman, M.J.; Kuchel, T.R.; Nash, C.H.; Edwards, J.; Bellomo, R. Structure and Function of the Kidney in Septic Shock. A Prospective Controlled Experimental Study. Am. J. Respir. Crit. Care Med. 2016, 194, 692–700.
- Di Giantomasso, D.; May, C.N.; Bellomo, R. Vital Organ Blood Flow During Hyperdynamic Sepsis. Chest 2003, 124, 1053–1059.
- Langenberg, C.; Gobe, G.; Hood, S.; May, C.N.; Bellomo, R. Renal Histopathology During Experimental Septic Acute Kidney Injury and Recovery. Crit. Care Med. 2014, 42, e58–e67.
- Ma, S.; Evans, R.; Iguchi, N.; Tare, M.; Parkington, H.C.; Bellomo, R.; May, C.N.; Lankadeva, Y.R. Sepsis-induced acute kidney injury: A disease of the microcirculation. Microcirculation 2018, 26, e12483.
- Calzavacca, P.; Evans, R.G.; Bailey, M.; Bellomo, R.; May, C.N. Cortical and Medullary Tissue Perfusion and Oxygenation in Experimental Septic Acute Kidney Injury. Crit. Care Med. 2015, 43, e431–e439.
- Lankadeva, Y.R.; Kosaka, J.; Evans, R.G.; Bellomo, R.; May, C.N. Urinary oxygenation as a surrogate marker of medullary oxy-genation during angiotensin II therapy in septic acute kidney injury. Crit. Care Med. 2018, 46, e41–e48.
- Lankadeva, Y.R.; Kosaka, J.; Evans, R.G.; Bailey, M.; Bellomo, R.; May, C.N. Intra-renal and urinary oxygenation during norepinephrine resuscitation in ovine septic acute kidney injury. Kidney Int. 2016, 90, 100–108.
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95.
- Kellum, J.A.; Prowle, J. Paradigms of acute kidney injury in the intensive care setting. Nat. Rev. Nephrol. 2018, 14, 217–230.
- Gomez, H.; Ince, C.; De Backer, D.; Pickkers, P.; Payen, D.; Hotchkiss, J.; Kellum, J.A. A unified theory of sepsis-induced acute kidney injury: Inflammation, microcirculatory dysfunction, bioenergetics and the tubular cell adaptation to injury. Shock 2014, 41, 3–11.
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167.
- Graham, D.B.; Robertson, C.M.; Bautista, J.; Mascarenhas, F.; Diacovo, M.J.; Montgrain, V.; Lam, S.K.; Cremasco, V.; Dunne, W.M.; Faccio, R.; et al. Neutrophil-mediated oxidative burst and host defense are controlled by a Vav-PLCgamma2 signaling axis in mice. J. Clin. Investig. 2007, 117, 3445–3452.
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell. Infect. Microbiol. 2017, 7, 373.
- Herter, J.M.; Rossaint, J.; Spieker, T.; Zarbock, A. Adhesion Molecules Involved in Neutrophil Recruitment during Sepsis-Induced Acute Kidney Injury. J. Innate Immun. 2014, 6, 597–606.
- Fujimi, S.; Ogura, H.; Tanaka, H.; Koh, T.; Hosotsubo, H.; Nakamori, Y.; Kuwagata, Y.; Shimazu, T.; Sugimoto, H. Activated Polymorphonuclear Leukocytes Enhance Production of Leukocyte Microparticles with Increased Adhesion Molecules in Patients with Sepsis. J. Trauma Acute Care Surg. 2002, 52, 443–448.
- Ware, L.B.; Fessel, J.P.; May, A.K.; Roberts, L.J. Plasma Biomarkers of Oxidant Stress and Development of Organ Failure in Severe Sepsis. Shock 2011, 36, 12–17.
- Satriano, J.A.; Banas, B.; Luckow, B.; Nelson, P.; Schlondorff, D.O. Regulation of RANTES and ICAM-1 expression in murine mesangial cells. J. Am. Soc. Nephrol. 1997, 8, 596–603.
- Ley, K.; Laudanna, C.; Cybulsky, M.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689.
- Michie, H.R.; Manogue, K.R.; Spriggs, D.R.; Revhaug, A.; O’Dwyer, S.; Dinarello, C.A.; Cerami, A.; Wolff, S.M.; Wilmore, D.W. Detection of Circulating Tumor Necrosis Factor after Endotoxin Administration. N. Engl. J. Med. 1988, 318, 1481–1486.
- Cannon, J.G.; Tompkins, R.G.; Gelfand, J.A.; Michie, H.R.; Stanford, G.G.; van der Meer, J.W.; Endres, S.; Lonnemann, G.; Corsetti, J.; Chernow, B.; et al. Circulating interleukin-1 and tumor necrosis factor in septic shock and experimental endotoxin fever. J. Infect. Dis. 1990, 161, 79–84.
- Chen, Y.; Jin, S.; Teng, X.; Hu, Z.; Zhang, Z.; Qiu, X.; Tian, D.; Wu, Y. Hydrogen Sulfide Attenuates LPS-Induced Acute Kidney Injury by Inhibiting Inflammation and Oxidative Stress. Oxidative Med. Cell. Longev. 2018, 2018, 6717212.
- Ehling, J.L.A.; Babickova, J.; Gremse, F.; Klinkhammer, B.M.; Baetke, S.C.; Knuechel, R.; Kiessling, F.; Floege, J.; Lammers, T.; Boor, P. Quantitative Micro-Computed Tomography Imaging of Vascular Dysfunction in Progressive Kidney Diseases. J. Am. Soc. Nephrol. 2016, 27, 520–532.
- Bábíčková, J.; Klinkhammer, B.M.; Buhl, E.M.; Djudjaj, S.; Hoss, M.; Heymann, F.; Tacke, F.; Floege, J.; Becker, J.U.; Boor, P. Regardless of etiology, progressive renal disease causes ultrastructural and functional alterations of peritubular capillaries. Kidney Int. 2017, 91, 70–85.
- Trzeciak, S.; Dellinger, R.P.; Parrillo, J.E.; Guglielmi, M.; Bajaj, J.; Abate, N.L.; Arnold, R.C.; Colilla, S.; Zanotti, S.; Hollenberg, S.M. Early microcirculatory perfusion derangements in patients with severe sepsis and septic shock: Relationship to hemody-namics, oxygen transport, and survival. Ann. Emerg. Med. 2007, 49, 88–98.
- McNeill, E.; Channon, K.M. The role of tetrahydrobiopterin in inflammation and cardiovascular disease. Thromb. Haemost. 2012, 108, 832–839.
- Bendall, J.K.; Alp, N.J.; Warrick, N.; Cai, S.; Adlam, D.; Rockett, K.; Yokoyama, M.; Kawashima, S.; Channon, K.M. Stoichi-ometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthase (eNOS) activity, and eNOS coupling In Vivo: Insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ. Res. 2005, 97, 864–871.
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: Implications for uncoupling endothelial nitric-oxide synthase. J. Biol. Chem. 2003, 278, 22546–22554.
- Alkaitis, M.S.; Crabtree, M.J. Recoupling the Cardiac Nitric Oxide Synthases: Tetrahydrobiopterin Synthesis and Recycling. Curr. Heart Fail. Rep. 2012, 9, 200–210.
- Kietadisorn, R.; Juni, R.P.; Moens, A.L. Tackling endothelial dysfunction by modulating NOS uncoupling: New insights into its pathogenesis and therapeutic possibilities. Am. J. Physiol. Metab. 2012, 302, E481–E495.
- Lankadeva, Y.R.; Singh, R.R.; Moritz, K.M.; Parkington, H.C.; Denton, K.M.; Tare, M. Renal Dysfunction Is Associated with a Reduced Contribution of Nitric Oxide and Enhanced Vasoconstriction After a Congenital Renal Mass Reduction in Sheep. Circulation 2015, 131, 280–288.
- He, X.; Su, F.; Velissaris, D.; Salgado, D.R.; de Souza Barros, D.; Lorent, S.; Taccone, F.S.; Vincent, J.L.; De Backer, D. Admin-istration of tetrahydrobiopterin improves the microcirculation and outcome in an ovine model of septic shock. Crit. Care Med. 2012, 40, 2833–2840.
- Chelazzi, C.; Villa, G.; Mancinelli, P.; De Gaudio, A.R.; Adembri, C. Glycocalyx and sepsis-induced alterations in vascular permeability. Crit. Care 2015, 19, 1–7.
- Clapp, B.R.; Hingorani, A.D.; Kharbanda, R.K.; Mohamed-Ali, V.; Stephens, J.W.; Vallance, P.; MacAllister, R.J. Inflamma-tion-induced endothelial dysfunction involves reduced nitric oxide bioavailability and increased oxidant stress. Cardiovasc. Res. 2004, 64, 172–178.
- Lankadeva, Y.R.; Okazaki, N.; Evans, R.G.; Bellomo, R.; May, C.N. Renal Medullary Hypoxia: A New Therapeutic Target for Septic Acute Kidney Injury? Semin. Nephrol. 2019, 39, 543–553.
- Evans, R.G.; Lankadeva, Y.R.; Cochrane, A.D.; Marino, B.; Iguchi, N.; Zhu, M.Z.L.; Hood, S.G.; Smith, J.A.; Bellomo, R.; Gardiner, B.S.; et al. Renal haemodynamics and oxygenation during and after cardiac surgery and cardiopulmonary bypass. Acta Physiol. 2018, 222, e12995.
- Lankadeva, Y.R.; Cochrane, A.D.; Marino, B.; Iguchi, N.; Hood, S.G.; Bellomo, R.; May, C.N.; Evans, R.G. Strategies that improve renal medullary oxygenation during experimental cardiopulmonary bypass may mitigate postoperative acute kidney injury. Kidney Int. 2019, 95, 1338–1346.
- Heyman, S.N.; Reichman, J.; Brezis, M. Pathophysiology of radiocontrast nephropathy: A role for medullary hypoxia. Investig. Radiol. 1999, 34, 685–691.
- Tanaka, S.; Tanaka, T.; Nangaku, M. Hypoxia as a key player in the AKI-to-CKD transition. Am. J. Physiol. Physiol. 2014, 307, F1187–F1195.
- Ullah, M.; Basile, D.P. Role of Renal Hypoxia in the Progression From Acute Kidney Injury to Chronic Kidney Disease. Semin. Nephrol. 2019, 39, 567–580.
- Evans, R.G.; Smith, D.W.; Lee, C.J.; Ngo, J.P.; Gardiner, B.S. What makes the kidney susceptible to hypoxia? Anat. Rec. 2020, 303, 2544–2552.
- Evans, R.G.; Ince, C.; Joles, J.A.; Smith, D.W.; May, C.N.; O’Connor, P.M.; Gardiner, B. Haemodynamic influences on kidney oxygenation: Clinical implications of integrative physiology. Clin. Exp. Pharmacol. Physiol. 2013, 40, 106–122.
- Calzavacca, P.; Evans, R.; Bailey, M.; Lankadeva, Y.R.; Bellomo, R.; May, C.N. Long-term measurement of renal cortical and medullary tissue oxygenation and perfusion in unanesthetized sheep. Am. J. Physiol. Integr. Comp. Physiol. 2015, 308, R832–R839.
- Bonventre, J.V.; Weinberg, J.M. Recent Advances in the Pathophysiology of Ischemic Acute Renal Failure. J. Am. Soc. Nephrol. 2003, 14, 2199–2210.
- Evans, R.G.; Gardiner, B.S.; Smith, D.W.; O’Connor, P.M. Intrarenal oxygenation: Unique challenges and the biophysical basis of homeostasis. Am. J. Physiol. Physiol. 2008, 295, F1259–F1270.
- Parikh, S.M.; Yang, Y.; He, L.; Tang, C.; Zhan, M.; Dong, Z. Mitochondrial Function and Disturbances in the Septic Kidney. Semin. Nephrol. 2015, 35, 108–119.
- Ince, C.; Mik, E.G. Microcirculatory and mitochondrial hypoxia in sepsis, shock, and resuscitation. J. Appl. Physiol. 2016, 120, 226–235.
- Bar-Or, D.; Carrick, M.M.; Mains, C.W.; Rael, L.T.; Slone, D.; Brody, E.N. Sepsis, oxidative stress, and hypoxia: Are there clues to better treatment? Redox Rep. 2015, 20, 193–197.
- Nagar, H.; Piao, S.; Kim, C.-S. Role of Mitochondrial Oxidative Stress in Sepsis. Acute Crit. Care 2018, 33, 65–72.
- Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807–819.
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138.
- Hernansanz-Agustín, P.; Ramos, E.; Navarro, E.; Parada, E.; Sánchez-López, N.; Peláez-Aguado, L.; Cabrera-García, J.D.; Tello, D.; Buendia, I.; Marina, A.; et al. Mitochondrial complex I deactivation is related to superoxide production in acute hypoxia. Redox Biol. 2017, 12, 1040–1051.
- Sureshbabu, A.; Patino, E.; Ma, K.C.; Laursen, K.; Finkelsztein, E.J.; Akchurin, O.; Muthukumar, T.; Ryter, S.W.; Gudas, L.; Choi, A.M.K.; et al. RIPK3 promotes sepsis-induced acute kidney injury via mitochondrial dysfunction. JCI Insight. 2018, 3, e98411.
- Kitur, K.; Wachtel, S.; Brown, A.; Wickersham, M.; Paulino, F.; Peñaloza, H.F.; Soong, G.; Bueno, S.; Parker, D.; Prince, A. Necroptosis Promotes Staphylococcus aureus Clearance by Inhibiting Excessive Inflammatory Signaling. Cell Rep. 2016, 16, 2219–2230.
- Duprez, L.; Takahashi, N.; Van Hauwermeiren, F.; Vandendriessche, B.; Goossens, V.; Berghe, T.V.; Declercq, W.; Libert, C.; Cauwels, A.; Vandenabeele, P. RIP Kinase-Dependent Necrosis Drives Lethal Systemic Inflammatory Response Syndrome. Immunity 2011, 35, 908–918.
- Brealey, D.; Brand, M.; Hargreaves, I.; Heales, S.; Land, J.; Smolenski, R.; Davies, N.A.; Cooper, C.; Singer, M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002, 360, 219–223.
- Plotnikov, E.Y.; Pevzner, I.B.; Zorova, L.D.; Chernikov, V.P.; Prusov, A.N.; Kireev, I.I.; Silachev, D.N.; Skulachev, V.P.; Zorov, D.B. Mitochondrial damage and mitochondria-Targeted antioxidant protection in LPS-induced acute kidney injury. Antioxidants 2019, 8, 176.
- Cain, K.; Bratton, S.B.; Cohen, G.M. The Apaf-1 apoptosome: A large caspase-activating complex. Biochimie 2002, 84, 203–214.
- Yuan, S.; Akey, C.W. Apoptosome structure, assembly, and procaspase activation. Structure 2013, 21, 501–515.
- Van der Slikke, E.C.; Star, B.S.; van Meurs, M.; Henning, R.H.; Moser, J.; Bouma, H.R. Sepsis is associated with mitochondrial DNA damage and a reduced mitochondrial mass in the kidney of patients with sepsis-AKI. Crit. Care 2021, 25, 1–13.


