Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jun Sung Moon | + 2937 word(s) | 2937 | 2021-08-09 08:32:16 | | | |
| 2 | Lily Guo | Meta information modification | 2937 | 2021-09-08 03:26:30 | | | | |
| 3 | Lily Guo | Meta information modification | 2937 | 2021-09-08 03:27:03 | | | | |
| 4 | Dean Liu | Meta information modification | 2937 | 2021-09-28 06:03:46 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Moon, J.S. CD36 in Pancreatic β-Cell Pathophysiology. Encyclopedia. Available online: https://encyclopedia.pub/entry/13984 (accessed on 07 February 2026).
Moon JS. CD36 in Pancreatic β-Cell Pathophysiology. Encyclopedia. Available at: https://encyclopedia.pub/entry/13984. Accessed February 07, 2026.
Moon, Jun Sung. "CD36 in Pancreatic β-Cell Pathophysiology" Encyclopedia, https://encyclopedia.pub/entry/13984 (accessed February 07, 2026).
Moon, J.S. (2021, September 08). CD36 in Pancreatic β-Cell Pathophysiology. In Encyclopedia. https://encyclopedia.pub/entry/13984
Moon, Jun Sung. "CD36 in Pancreatic β-Cell Pathophysiology." Encyclopedia. Web. 08 September, 2021.
Copy Citation
CD36 is a transmembrane glycoprotein found in platelets, mononuclear phagocytes, adipocytes, hepatocytes, myocytes, taste bud cells, and a variety of other cell types.
CD36 in Pancreatic β-Cell Pathophysiology
1. Introduction
CD36 is a transmembrane glycoprotein found in platelets, mononuclear phagocytes, adipocytes, hepatocytes, myocytes, taste bud cells, and a variety of other cell types. It plays a role in lipid accumulation, inflammatory signaling, energy reprogramming, oxidative stress, and apoptosis, all of which contribute to metabolic dysfunction [1][2][3][4][5]. Many physiological and pathological factors, such as long-chain fatty acids and proteins containing thrombospondin structural homology domains and oxidized phospholipids, including oxidized LDL (oxLDL), perturb CD36 function and induce metabolic diseases [6]. The scavenger receptor functions of CD36 have been widely researched due to the possibility of CD36 ligands in disease etiology. For example, higher sCD36, a non-cell-bound CD36 found in human plasma that indirectly reflects CD36 expression in tissues, has been linked to obesity, insulin resistance, and diabetes according to recent community-based cohort research [7][8][9]. Yang et al. also found that inhibiting the integral membrane protein CD36 with pharmacological agents lowers body weight growth and improves glucose tolerance [10]. Furthermore, a rise in CD36 levels contributes to the advancement of obesity-related metabolic dysfunctions by increasing lipid accumulation and inflammation. These studies also showed that CD36 is a critical player in the development of obesity and type 2 diabetes caused by a high-fat diet [11]. Despite the fact that the pathophysiology of these metabolic illnesses is complicated, there is evidence that CD36 is implicated in the abnormal signaling and tissue damage observed in this context. Since CD36 is expressed in a variety of tissues, it is impossible to discuss every aspect within the scope of this review. In this review, we look at the molecular links between CD36 and metabolic disease in a broader framework, focusing on pancreatic β-cells and other cell systems during the progression of metabolic disorders.
2. Role of CD36 in Pancreatic β-Cell Pathophysiology
2.1. Glucotoxicity
CD36 exerts fundamental biological functions at the cellular and tissue levels in multiple homeostatic and pathological processes by its distinct binding sites. Pancreatic β-cells play a central role in regulating glucose metabolism to sustain energy homeostasis by mediating a balance between insulin, an anabolic hormone, and glucagon, a catabolic hormone. Pancreatic β-cells require suitable sensors and signaling molecules that are integrated to modulate insulin secretion and maintain homeostasis. However, type 2 diabetes (T2D) is based on the inability of pancreatic β-cells to sustain a compensatory secretory response, leading to insulin secretory dysfunction and the pathogenesis of T2D. CD36 is the most generously expressed transporter among fatty acid transporters in human islets. It is located in the plasma membrane and co-localizes with insulin granules [12]. Interestingly, CD36 was shown to traffic between intracellular compartments and the cell surface in a vesicle-mediated process [13]. It has been well established that glucose potentiates fatty acid-induced β-cell death via apoptosis [14][15]. Similarly, the overexpression of CD36 in β-cells increases the uptake of fatty acids and leads to metabolic and functional dysfunction [16]. We also reported that glucotoxicity influences pancreatic β-cell dysfunction by increasing the influx of free fatty acids (FFAs) via CD36 [17]. To evaluate the mechanisms by which glucotoxicity affects β-cell dysfunction, we investigated CD36 expression and trafficking in β-cells and observed that Rac1, a small Rho family protein, displays increased glucose-mediated CD36 expression on the membrane surface in pancreatic β-cells [18]. The importance of Rac1 signaling in early-phase insulin secretion was previously demonstrated in β-cell-specific Rac1-deficient mice via the inhibition of F-actin depolymerization [19][20]. Glucose stimulates the recruitment of insulin granules to the cell membrane through actin remodeling, which is necessary for glucose-stimulated insulin secretion [21][22]. The F-actin function is coupled to SNARE-associated proteins such as syntaxin 1, syntaxin 4, and SNAP-25 in β-cells, and many F-actin-binding proteins interact with the SNARE machinery [21][23][24][25]. Several studies have shown that deficiencies in SNARE proteins are likely caused by high glucose and might contribute to cell dysfunction in disease states [26][27][28]. More considerations concerning SNARE protein function are discussed in the review article by Gaisano et al. [29]. Recently, it was shown that CD36 overexpression attenuates insulin secretion in human islets through the reduction of the exocytotic genes Snap25 and Vamp2, resulting in a decreased number of docked granules. It was further demonstrated that CD36 overexpression attenuates insulin signaling, resulting in the accumulation of the transcription factor FoxO1 in the nucleus as a potent transcriptional repressor of exocytotic genes. Interestingly, the inhibition of CD36 was shown to upregulate exocytotic gene expression in human islets, improving granule docking and resulting in increased insulin secretion without affecting insulin content [30]. Further research is required to identify the roles of CD36 in exocytotic gene function as well as whether F-actin function is involved in suppressing insulin secretion by CD36. Such studies will provide greater insights into the mechanisms of how CD36 induces metabolic dysfunction in pancreatic β-cells.
On the other hand, evidence suggests that hyperglycemia leads to the generation of reactive oxygen species (ROS), resulting in increased oxidative stress in β-cells [31][32]. The activation of Rac1 increases the production of oxidants, such as H2O2, via the activation of NADPH oxidase (NOX), which might trigger oxidative stress linked to β-cell death in T2D [33]. It was previously observed that CD36 deficiency reduces NOX activity and attenuates obesity-associated oxidative stress in the heart [34]. We also observed that Rac1 mediates NOX activity, leading to an increase in CD36 at the plasma membrane and that Rac1 and NOX inhibition can abrogate CD36 downstream signaling damage in response to high glucose [18]. However, it remains unknown how CD36 translocation to the plasma membrane is detected after Rac1-NOX activation by high glucose. One possible explanation may involve the palmitoylation of CD36 by supraphysiologic glucose levels. High-glucose-induced Rac1-palmitoylation has been suggested to be a driving force behind the activation of NOX, which in turn would alter the localization of Rac1 remodeling in diabetic retinopathy [35]. Nonetheless, a current topic of research is to elucidate how protein palmitoylation influences the function of proteins under high glucose conditions in pancreatic β-cells. It should be noted that increasing ROS production can alter cellular dysfunction stimulated by the activation of stress kinases by changing the balance of antioxidant enzymes. Previous findings also reported that CD36 altered cellular signaling under metabolic stress conditions by downregulating the redox-sensitive nuclear factor Nrf2 via Fyn kinase in murine vascular smooth muscle cells [36]. In addition, CD36 signaling in response to scavenger ligands leads to the activation of Src and MAPK family kinases, such as Lyn and c-Jun N-terminal kinase (JNK) in macrophages and platelets, whereas Fyn and p38 are the primary mediators of endothelial cells [37][38][39]. We also reported that Rac1-CD36 signaling by high-glucose-induced JNK and p38MAPK activation and the inhibition of CD36 inhibition blocks high-glucose-induced oxidative stress. Lots of evidence has suggested that ER stress is linked to insulin resistance, and pancreatic β-cell ER expansion was detected in patients with T2D [40][41]. Cells activate adaptive, self-protective mechanisms in response to ER stress, which are collectively referred to as the ER stress response (also named UPR). These include enhanced ER size, increased ER folding capacity through the manipulation of chaperones and foldases, decreased biosynthetic load, and the increased clearance of unfolded proteins through the stimulation of ER-related degradation. When these systems fail to alleviate the stress, apoptosis is triggered. Subsequent work from our lab has demonstrated that chronic glucose exposure or thapsigargin treatment induces ER stress through the reduced expression and activity of insulin and PDX1 with CD36 induction. Inhibition of CD36 in β-cells by metformin treatment or by using CD36 siRNA was shown to prevent the generation of ER stress markers and stress kinase activation [42]. It is clear that signaling related to CD36 regulation and its dynamics has impacts on oxidative stress, and understanding this linkage warrants further investigation. Given that CD36 signaling is related to numerous pathological events, β-cell CD36 downstream targets need to be further investigated.
2.2. Lipotoxicity
Hyperglycemia with elevated FFAs plays a significant role in insulin resistance and β-cell dysfunction in T2D [43][44][45]. Many studies have shown that glucose enhances fatty acid-induced β-cell death via apoptosis [14][15]. Fatty acid–glucose balance is essential for maintaining normal β-cell function, but lipotoxicity-induced β-cell dysfunction occurs with increased ROS, ceramide and nitric oxide levels, and mitochondrial perturbations [46][47]. Studies in Zucker diabetic fatty (ZDF) rats, an obesity-induced diabetic animal model, confirmed FFA-induced ceramide accumulation leading to β-cell apoptosis [48]. Another study demonstrated that superoxide production was elevated in islets isolated from Zucker lean fatty (ZLF) and Zucker diabetic fatty (ZDF) rats in the presence of glucose [49]. The resting superoxide content of ZDF rat islets was higher than that of Zucker lean control islets and was accompanied by the alteration of mitochondrial morphology. The FFA-induced formation of ceramide also induces the generation of ROS and DNA fragmentation [50]. Collectively, oxidative stress and mitochondrial dysfunction result in endogenous antioxidant impairment. However, plasma from patients with obesity and T2D shows enhanced levels of ceramides, which may serve as biomarkers for the diagnosis and treatment of obesity and diabetes [51][52][53]. Based on its role in FFA uptake, our lab showed that CD36 might promote ceramide-induced β-cell dysfunction by the Src-mediated tyrosine phosphorylation of Vav, a guanine nucleotide exchange factor (GEF), and also elevate metabolic pathways via its GEFs activity [54]. Evidence suggests that saturated fatty acids impair insulin secretion and induce insulin resistance via Src signaling in T2D [55]. Accordingly, we hypothesize that the induction of CD36 could be recapitulated in cells with functional vav tyrosine phosphorylation by Src promoting Rac1 signaling that generates ROS by NOX. Our results indicate that CD36-mediated Src-Vav activation is necessary for optimal Rac1-NADPH-induced superoxide production. To determine whether or not CD36 is linked to Vav-Rac1-NOX activation, we performed pharmacological inhibition of Src activity or CD36 siRNA, which significantly reduced ceramide-induced RAC1-NOX and inhibited ROS formation [56]. Holzer et al. demonstrated that saturated fatty acids stimulate stress-signaling activation by Src via transfer to a membrane micro domain [57]. In addition, the FFA-mediated Src-dependent Vav phosphorylation coordinates the engagement of Rac1-NOX-JNK signaling, which contributes to insulin resistance, obesity, and the production of inflammatory cytokines [58][59]. In podocytes, CD36-dependent uptake of palmitic acid leads to impaired mitochondrial energy metabolism, the alteration of mitochondrial and ER morphology, increased levels of mitochondrial ROS, the depolarization of mitochondria, ATP depletion, and apoptosis [60][61][62].
A recent study showed that p66Shc mediates lipotoxicity-induced impaired metabolic changes that promote pancreatic β-cell dysfunction and apoptosis in diabetes [32]. Earlier studies have suggested that p66Shc serine36 phosphorylation by JNK leads to ROS production and cell death [63]. Activated JNK combined with p66Shc serine36 phosphorylation activation to induce mitochondrial ROS in response to CD36 signaling promote cellular dysfunction. Cells lacking this pathway, as a consequence of CD36 inhibition, significantly block ceramide-induced β-cell dysfunction. Evidence points to CD36 signaling-generated H2O2, which promotes cysteine sulfenylation, a post-translational modification important to the augmentation of platelet activation and aggregation [64]. Peroxiredoxins, a thioredoxin-dependent peroxide reductase family of antioxidant proteins, catalyze the reduction of both hydrogen peroxide and alkyl peroxides to water and their corresponding alcohols [65][66]. The expression of peroxiredoxin-3 (PRDX3) is restricted to β-cells in pancreatic tissue. The oxidation of peroxidase cysteine to sulfonic acid (peroxiredoxin-SO3) promotes the accumulation of oxidized PRDX3 in mitochondria, which favors mitochondrial permeability transition pore (MPTP) opening and mitochondrial swelling [67]. Importantly, ceramide-induced sulfenylation is reduced in the presence of CD36 inhibition, which is consistent with a CD36-dependent mechanism. However, reduced PRDX3 is regulated by the thioredoxin–thioredoxin reductase system. Accordingly, we hypothesize that the inhibition of thioredoxin could be recapitulated in cells with functional thioredoxin-interacting protein (TXNIP) by preventing peroxiredoxin-3 activity in response to ceramide. We observed that TXNIP translocates to mitochondria and inhibits the antioxidative protein thioredoxin in response to ceramide. Moreover, ceramide-induced nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation has been shown to increase TXNIP expression in β-cells [68]. This finding suggests that CD36 plays an important role in the initiation of oxidative stress induced by ceramide under conditions of β-cell failure. Thus, exploration of CD36 warrants further investigation.
Under normal conditions, mitochondria in β-cells persistently undergo fusion and fission. These processes may function to refute the negative impacts of the long-term presentation of β-cells to palmitate under high glucose conditions, causing mitochondrial fragmentation and impeding network dynamics by abolishing fusion and fission activity [69]. There are two powerfully contradictory processes that determine mitochondrial shape and morphology: fusion and fission. The ablation of both fusion and fission produces a significant effect on the progression of cells to apoptosis [70]. It has been reported that the mitochondria of β-cells from Zucker diabetic rats are divided, suggesting an imbalance in the mitochondrial fusion and fission process [49]. The exposure of β-cells to high-fat glucose conditions causes the discharge of Ca2+ from the ER to the cytoplasm, driving a rise in the cytosolic Ca2+ concentration that reflects expanded mitochondrial Ca2+ uptake. Increased mitochondrial Ca2+ uptake improves local buffering capacity and the discharge of proteins competent in apoptosis induction. Hence, Ca2+ activates the phosphatase calcineurin, which dephosphorylates and inactivates dynamin-related protein 1 (Drp1), a master controller of mitochondrial fission [71]. It can be assumed that crosstalk between the ER and mitochondria may promote cellular commitment to apoptosis through Ca2+. Recently, the presence of inositol trisphosphate receptor (IP3Rs) has been implicated in proapoptotic Ca2+ transfer between the ER and mitochondria [72]. An important remark is that Akt restrains the ER-to-mitochondria Ca2+ exchange by means of IP3R3 and ensures against Ca2+ intervened apoptosis [73]. Interestingly, CD36 was found to be overexpressed in obese diabetic islets and suppressed the insulin-signaling PI3K/AKT pathway, as well as its downstream transcription factors [30]. These effects result in ER-mitochondrial reprogramming, which contributes to the development of β-cell death and failure. The different pathways enacted by CD36 require further confirmation of the precise roles of CD36 signaling pathways in β-cell failure.
2.3. OX-LDL and Amyloid Deposition
CD36 can generate cell-specific reactions to multiple ligands through the binding of context-specific binding partners that contribute to the development of β-cell dysfunction. As described above, it has been shown that ER stress is connected to insulin resistance in diabetes, and, conjointly, an expansion of ER was recognized in β-cells from patients with T2D [39][40]. Oxidized-LDL (oxLDL)-induced ER stress activation is coupled with oxidative stress, leading to β-cell dysfunction and death [74]. It has been shown that oxLDL induces β-cell dysfunction and apoptosis via the activation of ROS and that radical lipid hydroperoxides contribute to JNK activation [75][76][77]. However, the downstream mechanism by which JNK leads to apoptosis is not yet clear, and the crosslink between oxLDL and CD36 may promote cellular commitment to apoptosis through JNK enactment. A previous study reported that oxLDL intervenes with the JNK-dependent phosphorylation of p66Shc in endothelial cells, which contributes to oxidative stress and the atherogenic progression [78], Thus, we cannot preclude a role for PRDX3 oxidation in CD36 signaling. In this way, p66Shc can result in the overproduction of H2O2, which in turn can react with PRDX3 to cause toxic mitochondrial dysfunction and apoptosis.
Regarding the molecular mechanisms involved, CD36 overexpression partners with the increased uptake of oxLDL without exerting additive effects on oxLDL toxicity [79]. Evidence suggests that CD36 causes a mitochondrial metabolic switch from oxidative phosphorylation to superoxide generation in reaction to oxLDL, which subsequently promotes NF-κB activation and the generation of pro-inflammatory cytokines [80]. Hence, redox status is subordinate on the degree to which a cell’s components exist in an oxidative state, whereby a reducing environment inside cells can prevent oxidative stress. oxLDL also initiated the ASM/ceramide signaling pathway, which is involved in macrophage apoptosis via the ER stress pathway [81]. However, the downstream targets of oxLDL in β-cells are not well known, and additional studies are needed.
On the other hand, β-cells have a lower abundance of antioxidant defense enzymes, such as superoxide dismutase (SOD), catalase, and glutathione peroxidase (GPx) [82][83][84]. As such, the administration of antioxidant supplements can increase the defense capacity of islet cells to cope with oxidative stress [85]. Vitamin E is a redox-active natural compound that downregulates levels of ROS under different experimental conditions [86][87][88][89]. Interestingly, vitamin E reduces the uptake of OX-LDL by inhibiting CD36 expression via the PPARγ signaling pathway [90][91][92]. Furthermore, vitamin E facilitates the activation of PI3Kγ/AKT, leading to increased VEGF expression as well as elevation of cell survival and angiogenesis via its ability to increase tissue remodeling [93]. In addition, genetic data indicate that VEGF is a major regulator of islet vascularization and the revascularization of transplanted islets, and reduced beta-cell VEGF expression impairs glucose-stimulated insulin secretion [94]. Furthermore, the addition of vitamin E induces insulin secretion and islet-cell survival and functionality by enhancing PDX1, a master regulator of insulin gene expression [95][96]. Thus, the elevated expression of CD36 in beta cells exposed to elevated OX-LDL results in increased ROS expression that could induce discrete oxidative stress. Therefore, we suggest that the vitamin E-induced elevation of insulin expression may be mediated by CD36 inhibition, which may explain, at least in part, the reported protection against oxidative stress. Further studies are needed to elucidate the relationship between CD36, vitamin E, and OX-LDL in the pancreatic β-cell dysfunction.
Among the variety of proapoptotic factors present in pancreatic β-cells, islet amyloid polypeptide (IAPP) is thought to play a crucial role in β-cell apoptosis. The proapoptotic effects of IAPP are mediated through a complex sequence of signaling events that lead to defects in mitochondrial dysfunction, autophagy, local inflammation, oxidative stress, cytokine production, and the enactment of signaling pathways driving to apoptosis [97][98][99][100]. Interestingly, CD36 can create a solid pro-inflammatory reaction through its interaction with secreted amyloid-beta 1–42 (Aβ) in macrophages [101]. However, the presence of this CD36-dependent pro-inflammatory signaling hub within the pancreatic β-cells has not been studied and warrants further investigation (Figure 1).
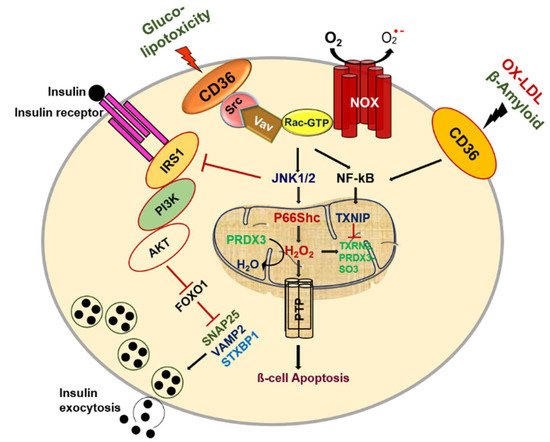
Figure 1. CD36 signal transduction in pancreatic β-cell dysfunction.
References
- Harmon, C.M.; Abumrad, N.A. Binding of sulfosuccinimidyl fatty acids to adipocyte membrane proteins: Isolation and amino-terminal sequence of an 88-kD protein implicated in transport of long-chain fatty acids. J. Membr. Biol. 1993, 133, 43–49.
- Fukuwatari, T.; Kawada, T.; Tsuruta, M.; Hiraoka, T.; Iwanaga, T.; Sugimoto, E.; Fushiki, T. Expression of the putative membrane fatty acid transporter (FAT) in taste buds of the circumvallate papillae in rats. FEBS Lett. 1997, 414, 461–464.
- Van Nieuwenhoven, F.A.; Verstijnen, C.P.; Abumrad, N.A.; Willemsen, P.H.; Van Eys, G.J.; Van der Vusse, G.J.; Glatz, J.F. Putative membrane fatty acid translocase and cytoplasmic fatty acid-binding protein are co-expressed in rat heart and skeletal muscles. Biochem. Biophys. Res. Commun. 1995, 207, 747–752.
- Savill, J.; Hogg, N.; Ren, Y.; Haslett, C. Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. J. Clin. Investig. 1992, 90, 1513–1522.
- Ghosh, A.; Li, W.; Febbraio, M.; Espinola, R.G.; McCrae, K.R.; Cockrell, E.; Silverstein, R.L. Platelet CD36 mediates interactions with endothelial cell-derived microparticles and contributes to thrombosis in mice. J. Clin. Investig. 2008, 118, 1934–1943.
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat. Med. 2007, 13, 1086–1095.
- Koonen, D.P.; Jensen, M.K.; Handberg, A. Soluble CD36- a marker of the (pathophysiological) role of CD36 in the metabolic syndrome? Arch. Physiol. Biochem. 2011, 117, 57–63.
- Wang, Y.; Koch, M.; di Giuseppe, R.; Evans, K.; Borggrefe, J.; Nöthlings, U.; Handberg, A.; Jensen, M.K.; Lieb, W. Associations of plasma CD36 and body fat distribution. J. Clin. Endocrinol. Metab. 2019, 104, 4016–4023.
- Kennedy, D.J.; Kashyap, S.R. Pathogenic role of scavenger receptor CD36 in the metabolic syndrome and diabetes. Metab. Syndr. Relat. Disord. 2011, 9, 239–245.
- Yang, J.; Park, K.W.; Cho, S. Inhibition of the CD36 receptor reduces visceral fat accumulation and improves insulin resistance in obese mice carrying the BDNF-Val66Metvariant. J. Biol. Chem. 2018, 293, 13338–13348.
- Liu, M.; Tso, P.; Woods, S.C. Receptor CD36 links a risk-associated allele to obesity and metabolic disorders. J. Biol. Chem. 2018, 293, 13349–13350.
- Noushmehr, H.; D’Amico, E.; Farilla, L.; Hui, H.; Wawrowsky, K.A.; Mlynarski, W.; Doria, A.; Abumrad, N.A.; Perfetti, R. Fatty acid translocase (FAT/CD36) is localized on insulin-containing granules in human pancreatic beta-cells and mediates fatty acid effects on insulin secretion. Diabetes 2005, 54, 472–481.
- Schwenk, R.W.; Holloway, G.P.; Luiken, J.J.; Bonen, A.; Glatz, J.F. Fatty acid transport across the cell membrane: Regulation by fatty acid transporters. Prostaglandins Leukot. Essent. Fatty Acids 2010, 82, 149–154.
- Maedler, K.; Oberholzer, J.; Bucher, P.; Spinas, G.A.; Donath, M.Y. Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes 2003, 52, 726–733.
- Maedler, K.; Spinas, G.A.; Dyntar, D.; Moritz, W.; Kaiser, N.; Donath, M.Y. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 2001, 50, 69–76.
- Wallin, T.; Ma, Z.; Ogata, H.; Jorgensen, I.H.; Iezzi, M.; Wang, H.; Wollheim, C.B.; Bjorklund, A. Facilitation of fatty acid uptake by CD36 in insulin-producing cells reduces fatty-acid-induced insulin secretion and glucose regulation of fatty acid oxidation. Biochim. Biophys. Acta 2010, 1801, 191–197.
- Kim, Y.W.; Moon, J.S.; Seo, Y.J.; Park, S.Y.; Kim, J.Y.; Yoon, J.S.; Lee, I.K.; Lee, H.W.; Won, K.C. Inhibition of fatty acid translocase cluster determinant 36 (CD36), stimulated by hyperglycemia, prevents glucotoxicity in INS-1 cells. Biochem. Biophys. Res. Commun. 2012, 420, 462–466.
- Elumalai, S.; Karunakaran, U.; Lee, I.K.; Moon, J.S.; Won, K.C. Rac1-NADPH oxidase signaling promotes CD36 activation under glucotoxic conditions in pancreatic beta cells. Redox Biol. 2017, 11, 126–134.
- Asahara, S.; Shibutani, Y.; Teruyama, K.; Inoue, H.Y.; Kawada, Y.; Etoh, H.; Matsuda, T.; Kimura-Koyanagi, M.; Hashimoto, N.; Sakahara, M.; et al. Ras-related C3 botulinum toxin substrate 1 (RAC1) regulates glucose-stimulated insulin secretion via modulation of F-actin. Diabetologia 2013, 56, 1088–1097.
- Li, J.; Luo, R.; Kowluru, A.; Li, G. Novel regulation by Rac1 of glucose- and forskolin-induced insulin secretion in INS-1 beta-cells. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E818–E827.
- Thurmond, D.C.; Gonelle-Gispert, C.; Furukawa, M.; Halban, P.A.; Pessin, J.E. Glucose-stimulated insulin secretion is coupled to the interaction of actin with the t-SNARE (target membrane soluble N-ethylmaleimide-sensitive factor attachment protein receptor protein) complex. Mol. Endocrinol. 2003, 17, 732–742.
- Tomas, A.; Yermen, B.; Min, L.; Pessin, J.E.; Halban, P.A. Regulation of pancreatic beta-cell insulin secretion by actin cytoskeleton remodelling: Role of gelsolin and cooperation with the MAPK signalling pathway. J. Cell Sci. 2006, 119, 2156–2167.
- Band, A.M.; Ali, H.; Vartiainen, M.K.; Welti, S.; Lappalainen, P.; Olkkonen, V.M.; Kuismanen, E. Endogenous plasma membrane t-SNARE syntaxin 4 is present in rab11 positive endosomal membranes and associates with cortical actin cytoskeleton. FEBS Lett. 2002, 531, 513–519.
- Jewell, J.L.; Luo, W.; Oh, E.; Wang, Z.; Thurmond, D.C. Filamentous actin regulates insulin exocytosis through direct interaction with Syntaxin 4. J. Biol. Chem. 2008, 283, 10716–10726.
- Rondas, D.; Tomas, A.; Soto-Ribeiro, M.; Wehrle-Haller, B.; Halban, P.A. Novel mechanistic link between focal adhesion remodeling and glucose-stimulated insulin secretion. J. Biol. Chem. 2012, 287, 2423–2436.
- Nagamatsu, S.; Nakamichi, Y.; Yamamura, C.; Matsushima, S.; Watanabe, T.; Ozawa, S.; Furukawa, H.; Ishida, H. Decreased expression of t-SNAR.E.; syntaxin 1, and SNAP-25 in pancreatic beta-cells is involved in impaired insulin secretion from diabetic GK rat islets: Restoration of decreased t-SNARE proteins improves impaired insulin secretion. Diabetes 1999, 48, 2367–2373.
- Thurmond, D.C.; Gaisano, H.Y. Recent Insights into Beta-cell Exocytosis in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1310–1325.
- Torrejon-Escribano, B.; Escoriza, J.; Montanya, E.; Blasi, J. Glucose-dependent changes in SNARE protein levels in pancreatic beta-cells. Endocrinology 2011, 152, 1290–1299.
- Gaisano, H.Y. Recent new insights into the role of SNARE and associated proteins in insulin granule exocytosis. Diabetes Obes. Metab. 2017, 19 (Suppl. 1), 115–123.
- Nagao, M.; Esguerra, J.L.S.; Asai, A.; Ofori, J.K.; Edlund, A.; Wendt, A.; Sugihara, H.; Wollheim, C.B.; Oikawa, S.; Eliasson, L. Potential Protection Against Type 2 Diabetes in Obesity Through Lower CD36 Expression and Improved Exocytosis in beta-Cells. Diabetes 2020, 69, 1193–1205.
- Cnop, M.; Welsh, N.; Jonas, J.C.; Jorns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: Many differences, few similarities. Diabetes 2005, 54 (Suppl. 2), S97–S107.
- Ihara, Y.; Toyokuni, S.; Uchida, K.; Odaka, H.; Tanaka, T.; Ikeda, H.; Hiai, H.; Seino, Y.; Yamada, Y. Hyperglycemia causes oxidative stress in pancreatic beta-cells of GK rats, a model of type 2 diabetes. Diabetes 1999, 48, 927–932.
- Guichard, C.; Moreau, R.; Pessayre, D.; Epperson, T.K.; Krause, K.H. NOX family NADPH oxidases in liver and in pancreatic islets: A role in the metabolic syndrome and diabetes? Biochem. Soc. Trans. 2008, 36, 920–929.
- Gharib, M.; Tao, H.; Fungwe, T.V.; Hajri, T. Cluster Differentiating 36 (CD36) Deficiency Attenuates Obesity-Associated Oxidative Stress in the Heart. PLoS ONE 2016, 11, e0155611.
- Veluthakal, R.; Kumar, B.; Mohammad, G.; Kowluru, A.; Kowluru, R.A. Tiam1-Rac1 Axis Promotes Activation of p38 MAP Kinase in the Development of Diabetic Retinopathy: Evidence for a Requisite Role for Protein Palmitoylation. Cell Physiol. Biochem. 2015, 36, 208–220.
- Li, W.; Febbraio, M.; Reddy, S.P.; Yu, D.Y.; Yamamoto, M.; Silverstein, R.L. CD36 participates in a signaling pathway that regulates ROS formation in murine VSMCs. J. Clin. Investig. 2010, 120, 3996–4006.
- Jimenez, B.; Volpert, O.V.; Crawford, S.E.; Febbraio, M.; Silverstein, R.L.; Bouck, N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat. Med. 2000, 6, 41–48.
- Moore, K.J.; El Khoury, J.; Medeiros, L.A.; Terada, K.; Geula, C.; Luster, A.D.; Freeman, M.W. A CD36-initiated signaling cascade mediates inflammatory effects of beta-amyloid. J. Biol. Chem. 2002, 277, 47373–47379.
- Rahaman, S.O.; Lennon, D.J.; Febbraio, M.; Podrez, E.A.; Hazen, S.L.; Silverstein, R.L. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006, 4, 211–221.
- Marchetti, P.; Bugliani, M.; Lupi, R.; Marselli, L.; Masini, M.; Boggi, U.; Filipponi, F.; Weir, G.C.; Eizirik, D.L.; Cnop, M. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia 2007, 50, 2486–2494.
- Nakatani, Y.; Kaneto, H.; Kawamori, D.; Yoshiuchi, K.; Hatazaki, M.; Matsuoka, T.A.; Ozawa, K.; Ogawa, S.; Hori, M.; Yamasaki, Y.; et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J. Biol. Chem. 2005, 280, 847–851.
- Moon, J.S.; Karunakaran, U.; Elumalai, S.; Lee, I.K.; Lee, H.W.; Kim, Y.W.; Won, K.C. Metformin prevents glucotoxicity by alleviating oxidative and ER stress-induced CD36 expression in pancreatic beta cells. J. Diabetes Complicat. 2017, 31, 21–30.
- Karunakaran, U.; Kim, H.J.; Kim, J.Y.; Lee, I.K. Guards and culprits in the endoplasmic reticulum: Glucolipotoxicity and beta-cell failure in type II diabetes. Exp. Diabetes Res. 2012, 2012, 639762.
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent Insights Into Mechanisms of beta-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1514–1534.
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: Fuel excess and beta-cell dysfunction. Endocr. Rev. 2008, 29, 351–366.
- Boslem, E.; Meikle, P.J.; Biden, T.J. Roles of ceramide and sphingolipids in pancreatic beta-cell function and dysfunction. Islets 2012, 4, 177–187.
- Maestre, I.; Jordan, J.; Calvo, S.; Reig, J.A.; Cena, V.; Soria, B.; Prentki, M.; Roche, E. Mitochondrial dysfunction is involved in apoptosis induced by serum withdrawal and fatty acids in the beta-cell line INS-1. Endocrinology 2003, 144, 335–345.
- Shimabukuro, M.; Ohneda, M.; Lee, Y.; Unger, R.H. Role of nitric oxide in obesity-induced beta cell disease. J. Clin. Investig. 1997, 100, 290–295.
- Shimabukuro, M.; Zhou, Y.T.; Levi, M.; Unger, R.H. Fatty acid-induced beta cell apoptosis: A link between obesity and diabetes. Proc. Natl. Acad. Sci. USA 1998, 95, 2498–2502.
- Bindokas, V.P.; Kuznetsov, A.; Sreenan, S.; Polonsky, K.S.; Roe, M.W.; Philipson, L.H. Visualizing superoxide production in normal and diabetic rat islets of Langerhans. J. Biol. Chem. 2003, 278, 9796–9801.
- Lupi, R.; Dotta, F.; Marselli, L.; Del Guerra, S.; Masini, M.; Santangelo, C.; Patane, G.; Boggi, U.; Piro, S.; Anello, M.; et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: Evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes 2002, 51, 1437–1442.
- Haus, J.M.; Kashyap, S.R.; Kasumov, T.; Zhang, R.; Kelly, K.R.; Defronzo, R.A.; Kirwan, J.P. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 2009, 58, 337–343.
- Samad, F.; Hester, K.D.; Yang, G.; Hannun, Y.A.; Bielawski, J. Altered adipose and plasma sphingolipid metabolism in obesity: A potential mechanism for cardiovascular and metabolic risk. Diabetes 2006, 55, 2579–2587.
- Wigger, L.; Cruciani-Guglielmacci, C.; Nicolas, A.; Denom, J.; Fernandez, N.; Fumeron, F.; Marques-Vidal, P.; Ktorza, A.; Kramer, W.; Schulte, A.; et al. Plasma Dihydroceramides Are Diabetes Susceptibility Biomarker Candidates in Mice and Humans. Cell Rep. 2017, 18, 2269–2279.
- Bustelo, X.R. Vav family exchange factors: An integrated regulatory and functional view. Small GTPases 2014, 5, 9.
- Kominato, R.; Fujimoto, S.; Mukai, E.; Nakamura, Y.; Nabe, K.; Shimodahira, M.; Nishi, Y.; Funakoshi, S.; Seino, Y.; Inagaki, N. Src activation generates reactive oxygen species and impairs metabolism-secretion coupling in diabetic Goto-Kakizaki and ouabain-treated rat pancreatic islets. Diabetologia 2008, 51, 1226–1235.
- Karunakaran, U.; Elumalai, S.; Moon, J.S.; Won, K.C. CD36 dependent redoxosomes promotes ceramide-mediated pancreatic beta-cell failure via p66Shc activation. Free Radic. Biol. Med. 2019, 134, 505–515.
- Holzer, R.G.; Park, E.J.; Li, N.; Tran, H.; Chen, M.; Choi, C.; Solinas, G.; Karin, M. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 2011, 147, 173–184.
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336.
- Kant, S.; Standen, C.L.; Morel, C.; Jung, D.Y.; Kim, J.K.; Swat, W.; Flavell, R.A.; Davis, R.J. A Protein Scaffold Coordinates SRC-Mediated JNK Activation in Response to Metabolic Stress. Cell Rep. 2017, 20, 2775–2783.
- Hua, W.; Huang, H.Z.; Tan, L.T.; Wan, J.M.; Gui, H.B.; Zhao, L.; Ruan, X.Z.; Chen, X.M.; Du, X.G. CD36 Mediated Fatty Acid-Induced Podocyte Apoptosis via Oxidative Stress. PLoS ONE 2015, 10, e0127507.
- Xu, S.; Nam, S.M.; Kim, J.H.; Das, R.; Choi, S.K.; Nguyen, T.T.; Quan, X.; Choi, S.J.; Chung, C.H.; Lee, E.Y.; et al. Palmitate induces ER calcium depletion and apoptosis in mouse podocytes subsequent to mitochondrial oxidative stress. Cell Death Dis. 2015, 6, e1976.
- Natalicchio, A.; Tortosa, F.; Labarbuta, R.; Biondi, G.; Marrano, N.; Carchia, E.; Leonardini, A.; Cignarelli, A.; Bugliani, M.; Marchetti, P.; et al. The p66(Shc) redox adaptor protein is induced by saturated fatty acids and mediates lipotoxicity-induced apoptosis in pancreatic beta cells. Diabetologia 2015, 58, 1260–1271.
- Khalid, S.; Drasche, A.; Thurner, M.; Hermann, M.; Ashraf, M.I.; Fresser, F.; Baier, G.; Kremser, L.; Lindner, H.; Troppmair, J. cJun N-terminal kinase (JNK) phosphorylation of serine 36 is critical for p66Shc activation. Sci. Rep. 2016, 6, 20930.
- Yang, M.; Li, W.; Harberg, C.; Chen, W.; Yue, H.; Ferreira, R.B.; Wynia-Smith, S.L.; Carroll, K.S.; Zielonka, J.; Flaumenhaft, R.; et al. Cysteine sulfenylation by CD36 signaling promotes arterial thrombosis in dyslipidemia. Blood Adv. 2020, 4, 4494–4507.
- Kang, S.W.; Chae, H.Z.; Seo, M.S.; Kim, K.; Baines, I.C.; Rhee, S.G. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpha. J. Biol. Chem. 1998, 273, 6297–6302.
- Lim, Y.S.; Cha, M.K.; Kim, H.K.; Uhm, T.B.; Park, J.W.; Kim, K.; Kim, I.H. Removals of hydrogen peroxide and hydroxyl radical by thiol-specific antioxidant protein as a possible role in vivo. Biochem. Biophys. Res. Commun. 1993, 192, 273–280.
- Cox, A.G.; Pullar, J.M.; Hughes, G.; Ledgerwood, E.C.; Hampton, M.B. Oxidation of mitochondrial peroxiredoxin 3 during the initiation of receptor-mediated apoptosis. Free Radic. Biol. Med. 2008, 44, 1001–1009.
- Karunakaran, U.; Moon, J.S.; Lee, H.W.; Won, K.C. CD36 initiated signaling mediates ceramide-induced TXNIP expression in pancreatic beta-cells. Biochim. Biophys. Acta 2015, 1852, 2414–2422.
- Park, K.S.; Wiederkehr, A.; Kirkpatrick, C.; Mattenberger, Y.; Martinou, J.C.; Marchetti, P.; Demaurex, N.; Wollheim, C.B. Selective actions of mitochondrial fission/fusion genes on metabolism-secretion coupling in insulin-releasing cells. J. Biol. Chem. 2008, 283, 33347–33356.
- Wasilewski, M.; Scorrano, L. The changing shape of mitochondrial apoptosis. Trends Endocrinol. Metab. 2009, 20, 287–294.
- Koshkin, V.; Dai, F.F.; Robson-Doucette, C.A.; Chan, C.B.; Wheeler, M.B. Limited mitochondrial permeabilization is an early manifestation of palmitate-induced lipotoxicity in pancreatic beta-cells. J. Biol. Chem. 2008, 283, 7936–7948.
- De Stefani, D.; Bononi, A.; Romagnoli, A.; Messina, A.; De Pinto, V.; Pinton, P.; Rizzuto, R. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 2012, 19, 267–273.
- Marchi, S.; Marinello, M.; Bononi, A.; Bonora, M.; Giorgi, C.; Rimessi, A.; Pinton, P. Selective modulation of subtype III IP(3)R by Akt regulates ER Ca(2)(+) release and apoptosis. Cell Death Dis. 2012, 3, e304.
- Plaisance, V.; Brajkovic, S.; Tenenbaum, M.; Favre, D.; Ezanno, H.; Bonnefond, A.; Bonner, C.; Gmyr, V.; Kerr-Conte, J.; Gauthier, B.R.; et al. Endoplasmic Reticulum Stress Links Oxidative Stress to Impaired Pancreatic Beta-Cell Function Caused by Human Oxidized LDL. PLoS ONE 2016, 11, e0163046.
- Abderrahmani, A.; Niederhauser, G.; Favre, D.; Abdelli, S.; Ferdaoussi, M.; Yang, J.Y.; Regazzi, R.; Widmann, C.; Waeber, G. Human high-density lipoprotein particles prevent activation of the JNK pathway induced by human oxidised low-density lipoprotein particles in pancreatic beta cells. Diabetologia 2007, 50, 1304–1314.
- Cnop, M.; Hannaert, J.C.; Grupping, A.Y.; Pipeleers, D.G. Low density lipoprotein can cause death of islet beta-cells by its cellular uptake and oxidative modification. Endocrinology 2002, 143, 3449–3453.
- Grupping, A.Y.; Cnop, M.; Van Schravendijk, C.F.; Hannaert, J.C.; Van Berkel, T.J.; Pipeleers, D.G. Low density lipoprotein binding and uptake by human and rat islet beta cells. Endocrinology 1997, 138, 4064–4068.
- Shi, Y.; Cosentino, F.; Camici, G.G.; Akhmedov, A.; Vanhoutte, P.M.; Tanner, F.C.; Luscher, T.F. Oxidized low-density lipoprotein activates p66Shc via lectin-like oxidized low-density lipoprotein receptor-1, protein kinase C-beta, and c-Jun N-terminal kinase kinase in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2090–2097.
- Ma, Z.; Ketelhuth, D.F.J.; Wirstrom, T.; Ohki, T.; Forteza, M.J.; Wang, H.; Grill, V.; Wollheim, C.B.; Bjorklund, A. Increased uptake of oxLDL does not exert lipotoxic effects in insulin-secreting cells. J. Mol. Endocrinol. 2019, 62, 159–168.
- Chen, Y.; Yang, M.; Huang, W.; Chen, W.; Zhao, Y.; Schulte, M.L.; Volberding, P.; Gerbec, Z.; Zimmermann, M.T.; Zeighami, A.; et al. Mitochondrial Metabolic Reprogramming by CD36 Signaling Drives Macrophage Inflammatory Responses. Circ. Res. 2019, 125, 1087–1102.
- Zhao, M.; Pan, W.; Shi, R.Z.; Bai, Y.P.; You, B.Y.; Zhang, K.; Fu, Q.M.; Schuchman, E.H.; He, X.X.; Zhang, G.G. Acid Sphingomyelinase Mediates Oxidized-LDL Induced Apoptosis in Macrophage via Endoplasmic Reticulum Stress. J. Atheroscler. Thromb. 2016, 23, 1111–1125.
- Benáková, Š.; Holendová, B.; Plecitá-Hlavatá, L. Redox Homeostasis in Pancreatic β-Cells: From Development to Failure. Antioxidants 2021, 10, 526.
- Ježek, P.; Holendová, B.; Jabůrek, M.; Tauber, J.; Dlasková, A.; Plecitá-Hlavatá, L. The Pancreatic β-Cell: The Perfect Redox System. Antioxidants 2021, 10, 197.
- Lenzen, S.; Drinkgern, J.; Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic. Biol. Med. 1996, 20, 463–466.
- Kaneto, H.; Kajimoto, Y.; Miyagawa, J.; Matsuoka, T.; Fujitani, Y.; Umayahara, Y.; Hanafusa, T.; Matsuzawa, Y.; Yamasaki, Y.; Hori, M. Beneficial effects of antioxidants in diabetes: Possible protection of pancreatic beta-cells against glucose toxicity. Diabetes 1999, 48, 2398–2406.
- Podszun, M.C.; Alawad, A.S.; Lingala, S.; Morris, N.; Huang, W.A.; Yang, S.; Schoenfeld, M.; Rolt, A.; Ouwerkerk, R.; Valdez, K.; et al. Vitamin E treatment in NAFLD patients demonstrates that oxidative stress drives steatosis through upregulation of de-novo lipogenesis. Redox Biol. 2020, 37, 101710.
- Ziegler, M.; Wallert, M.; Lorkowski, S.; Peter, K. Cardiovascular and Metabolic Protection by Vitamin E: A Matter of Treatment Strategy? Antioxidants 2020, 9, 935.
- Ungurianu, A.; Zanfirescu, A.; Nițulescu, G.; Margină, D. Vitamin E beyond Its Antioxidant Label. Antioxidants 2021, 10, 634.
- Di Vincenzo, A.; Tana, C.; El Hadi, H.; Pagano, C.; Vettor, R.; Rossato, M. Antioxidant, Anti-Inflammatory, and Metabolic Properties of Tocopherols and Tocotrienols: Clinical Implications for Vitamin E Supplementation in Diabetic Kidney Disease. Int. J. Mol. Sci. 2019, 20, 5101.
- Ozer, N.K.; Negis, Y.; Aytan, N.; Villacorta, L.; Ricciarelli, R.; Zingg, J.M.; Azzi, A. Vitamin E inhibits CD36 scavenger receptor expression in hypercholesterolemic rabbits. Atherosclerosis 2006, 184, 15–20.
- Ricciarelli, R.; Zingg, J.M.; Azzi, A. Vitamin E reduces the uptake of oxidized LDL by inhibiting CD36 scavenger receptor expression in cultured aortic smooth muscle cells. Circulation 2000, 102, 82–87.
- Munteanu, A.; Taddei, M.; Tamburini, I.; Bergamini, E.; Azzi, A.; Zingg, J.M. Antagonistic effects of oxidized low density lipoprotein and alpha-tocopherol on CD36 scavenger receptor expression in monocytes: Involvement of protein kinase B and peroxisome proliferator-activated receptor-gamma. J. Biol. Chem. 2006, 281, 6489–6497.
- Zingg, J.M.; Azzi, A.; Meydani, M. α-Tocopheryl Phosphate Induces VEGF Expression via CD36/PI3Kγ in THP-1 Monocytes. J. Cell Biochem. 2017, 118, 1855–1867.
- Brissova, M.; Shostak, A.; Shiota, M.; Wiebe, P.O.; Poffenberger, G.; Kantz, J.; Chen, Z.; Carr, C.; Jerome, W.G.; Chen, J.; et al. Pancreatic islet production of vascular endothelial growth factor—A is essential for islet vascularization, revascularization, and function. Diabetes 2006, 55, 2974–2985.
- Chia, L.L.; Jantan, I.; Chua, K.H. Tocotrienols Stimulate Insulin Secretion of Rat Pancreatic Isolated Islets in a Dynamic Culture. Curr. Pharm. Biotechnol. 2017, 18, 560–568.
- Hani, H.; Allaudin, Z.N.; Mohd-Lila, M.A.; Sarsaifi, K.; Rasouli, M.; Tam, Y.J.; Tengku-Ibrahim, T.A.; Othman, A.M. Improvement of isolated caprine islet survival and functionality in vitro by enhancing of PDX1 gene expression. Xenotransplantation 2017, 24, e12302.
- Abedini, A.; Schmidt, A.M. Mechanisms of islet amyloidosis toxicity in type 2 diabetes. FEBS Lett. 2013, 587, 1119–1127.
- Bishoyi, A.K.; Roham, P.H.; Rachineni, K.; Save, S.; Hazari, M.A.; Sharma, S.; Kumar, A. Human islet amyloid polypeptide (hIAPP)—A curse in type II diabetes mellitus: Insights from structure and toxicity studies. Biol. Chem. 2021, 402, 133–153.
- Subramanian, S.L.; Hull, R.L.; Zraika, S.; Aston-Mourney, K.; Udayasankar, J.; Kahn, S.E. cJUN N-terminal kinase (JNK) activation mediates islet amyloid-induced beta cell apoptosis in cultured human islet amyloid polypeptide transgenic mouse islets. Diabetologia 2012, 55, 166–174.
- Zhang, S.; Liu, J.; Dragunow, M.; Cooper, G.J. Fibrillogenic amylin evokes islet beta-cell apoptosis through linked activation of a caspase cascade and JNK1. J. Biol. Chem. 2003, 278, 52810–52819.
More
Information
Subjects:
Cell Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
884
Revisions:
4 times
(View History)
Update Date:
28 Sep 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No