 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Carmen Espinós | + 3155 word(s) | 3155 | 2021-08-30 08:33:19 | | | |
| 2 | Conner Chen | Meta information modification | 3155 | 2021-09-22 03:46:42 | | |
Video Upload Options
Wilson disease (WD) is a rare disorder caused by mutations in ATP7B, which leads to a defective biliary excretion of copper. Subsequent gradual accumulation of copper in different organs produces an extremely variable clinical picture, which comprises hepatic, neurological psychiatric, ophthalmological, and other disturbances. WD has specific treatment, so that early diagnosis is crucial in order to avoid the progression of the disease and its devastating consequences. The clinical diagnosis of WD is based on the Leipzig scale, which considers clinical, histological, biochemical and genetic data. However, even patients with an initial WD diagnosis based on a high Leipzig score result to suffer from other condition that mimics the WD’s phenotype (Wilson-like).
1. Introduction
Wilson's disease (WD; MIM 277900) is an autosomal recessive disorder related to the metabolism of copper, a metal that accumulates in tissues, mainly in liver and brain. WD is caused by mutations in the ATP7B gene, which encodes for a copper transporter that is responsible for biliary excretion of copper and its incorporation into ceruloplasmin. WD was first described by the British neurologist Samuel Alexander Kinnier Wilson as a " progressive lenticular degeneration accompanied by cirrhosis in the liver” [1]. The manuscript included four cases who presented with involuntary movements, spasticity, dysarthria, psychiatric disturbances and advanced cirrhosis. In the first half of the 20th century, several researchers made important contributions to the knowledge of the pathophysiological mechanisms of the disease. Rumpel reported excess copper in the liver of a patient with WD for the first time, and Cumings corroborated it and established that copper accumulation in the liver and in the basal ganglia were the etiological basis of the disease [2][3]. In less than 50 years since its discovery, WD became the first chronic hereditary liver disease that had a specific treatment, which made it possible to stop the devastating progression of the disease and prevent the eventual death. Despite these initial advances, diagnosis has remained a challenge and WD is currently an infradiagnosed disorder. Epidemiologic estimates have indeed established notable differences between clinical and genetic prevalence, which may be due to multiple factors including genetic modifiers, epigenetics and habits. The main underlying problem lies in suboptimal diagnostic tests and elusive biomarkers to diagnose a rare genetic disease characterized by incomplete penetrance and highly variable expression.
2. Clinical picture of Wilson’s disease
Wilson’s disease (WD) is a multisystemic disorder characterized by abnormal deposits of copper. Clinical signs depend on the organs where the copper is accumulated over the course of the pathological process (Figure 1). Accordingly, WD can present as a liver, neurological or mixed disease. Most pediatric patients usually exhibit a liver presentation, whereas patients diagnosed in adulthood predominantly manifest with a mixed presentation. While diagnosis is typically made in childhood, adolescence or early adulthood, between 5 and 35 years old, late presentations have been described.
Treatment is life-long and should never be interrupted. In most patients, a favorable response is reached with normalization of liver enzymes within 2-12 months from treatment initiation. Improvement of neurological manifestations may take longer up to 2-3 years. Treatment is divided into 2 phases, one initial decoppering phase and a second maintenance phase. Treatment is currently based on chelators (D-penicillamine or trientine) which promote renal copper excretion and zinc salts which block intestinal copper absorption and also stimulate cytoplasmic production of metallothionein, a protein that fixes copper in the cytoplasm of hepatocytes in a non-toxic way [4]. In some severe neurological cases, ammonium tetrathiomolybdate, an experimental drug, has been used for short periods of time. The choice of drug depends on the type and severity of clinical presentation, as well as center experience [5]. Newer formulations of trientine and tetrathiomolybdate are currently being evaluated in international trials [6]. In addition, gene therapy, which has shown to be effective in animal studies [7], is also entering the human arena.
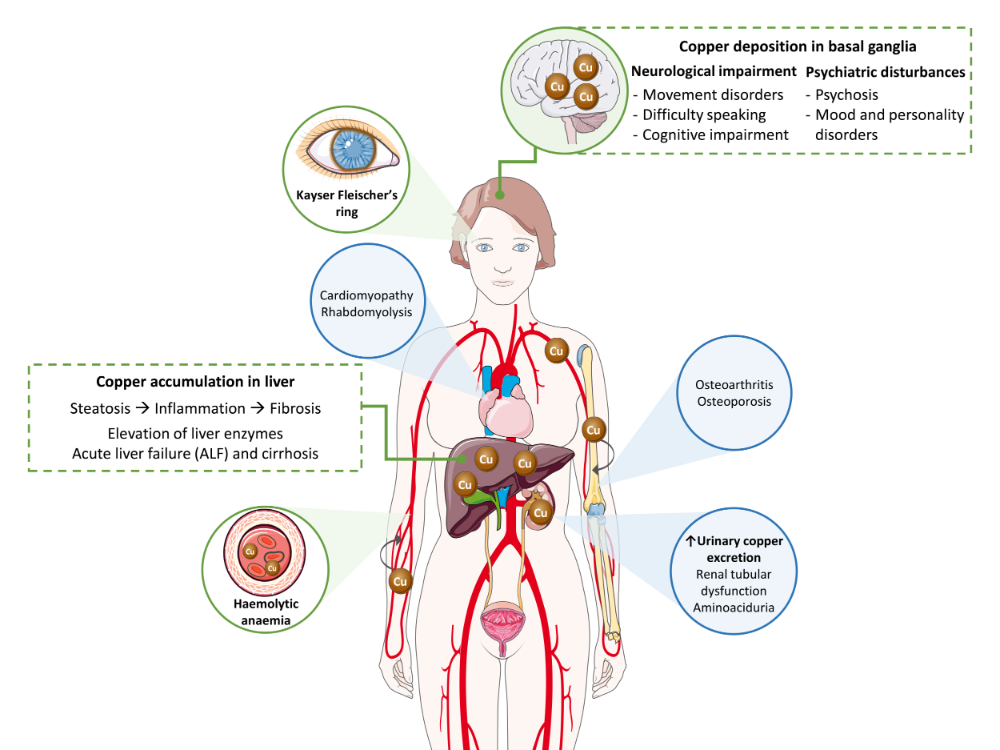 Figure 1. Copper toxicity in the pathogenesis of Wilson’s disease. Main signs and symptoms observed in patients are included in green boxes and circles, while secondary findings are contained in blue circles.
Figure 1. Copper toxicity in the pathogenesis of Wilson’s disease. Main signs and symptoms observed in patients are included in green boxes and circles, while secondary findings are contained in blue circles.
2.1. Clinical signs
2.1.1. Liver disease
Liver damage ranges from steatosis to chronic hepatitis, fibrosis and cirrhosis. Clinically it may present as asymptomatic hypertransaminasemia, with any of the manifestations of liver cirrhosis, or as severe acute liver failure (ALF). ALF may be the initial manifestation of latent WD or may develop weeks to months following treatment non-compliance. Although most patients have an underlying cirrhosis, the clinical picture is indistinguishable from that observed in the course of viral or toxic hepatitis. Urgent liver transplantation is generally required in these cases. Although not always present, Coombs negative hemolytic anemia, Kayser-Fleischer (KF) ring (present in about 50%), an AST/ALT ratio above 2 and low alkaline phosphatases serum levels are findings that help in reaching a diagnosis in patients with severe acute presentation.
2.1.2. Neurological and psychiatric disease
Neurological presentation is rarely seen in childhood, but may present in adolescence or early adulthood. It encompasses a wide spectrum of movement disorders caused by the degeneration of the basal ganglia (Figure 1) [8]. The more common are tremor, dystonia, parkinsonism, dysarthria, drooling and/or dysphagia. Moreover, additional features may also happen such as cerebellar dysfunction, chorea, hyperreflexia, seizures, restless legs syndrome, sleep disturbances, myoclonus and cognitive impairment [9]. Psychiatric symptoms secondary to neurological deterioration tend to go unnoticed in the early stages of disease. In adolescents, behavioural changes, emotional instability and poor academic performance are relatively common, while adult patients may show personality disorders, depression, anxiety, psychosis, and schizophrenia [10].
2.1.3. Additional clinical manifestations
When the copper concentration is excessive, this metal is released to the bloodstream in its free form, not bound to ceruloplasmin (NCC; non ceruloplasmin bound copper), and then, is deposited in other organs (Figure 1). A relevant ophthalmologic feature of WD is the KF ring, a greenish-brown opacity at the periphery of each iris. KF ring is detected in more than 90% of patients with neurological and/or psychiatric presentation, but only in ~50% of patients with liver symptoms, and rarely in those asymptomatic [10].
Excess of copper can lead to additional clinical features in other organs, including renal, osseomuscular, endocrine and cardiac manifestations, although these are rarer signs. Finally, other rare clinical manifestations include hypoparathyroidism, pancreatitis, amenorrhea and recurrent miscarriages [9].
3. Diagnosis: just a matter of the Leipzig scale?
WD clinical manifestations are diverse and not always present, potentially mimicking other diseases. In addition, none of the laboratory markers indicative of copper accumulation are 100% sensible and specific of the disease; hence, the need to combine clinical findings and biochemical parameters to reach a diagnosis. The diagnosis of WD is currently based on the score developed at the 8th International Meeting on Wilson Disease in Leipzig [11], which includes clinical signs, histopathological studies, biochemical tests and genetic analysis. A score ≥4 points would be confirmatory of WD, and this is already achievable if two deleterious mutations in ATP7B are detected segregating with the disease. Unfortunately, the clinical assessment and other tests included in the Leipzig score not always yield clear findings, and genetic testing is currently not available in all settings. Below we describe the tests commonly used in the diagnosis of WD, which are further discussed in [12].
Ceruloplasmin
Ceruloplasmin (Cp) is a copper-binding protein responsible for 75-90% transport of this metal in blood. When recently synthetized by hepatocytes, it is named apoceruloplasmin (Apo-Cp) and can bind six to eight copper ions, thus turning into its active form, holoceruloplasmin (Holo-Cp). ATP7B is the main protein which “shuttles” copper to Apo-Cp, and if this event does not occur, Apo-Cp will be rapidly degraded due to its low stability.
To measure circulating Cp levels, immunoassays are the most extended in clinical laboratories; yet, they can yield overestimated values, since both Apo-Cp and Holo-Cp are detected. Reference values for immunoassays vary between 0.15 and 0.20 g/L; levels below 0.20 g/L are suggestive but if lower than 0.10 g/L, a WD diagnosis is probable [11].
Cp levels tend to increase in case of inflammation, infection and hyperestrogenemia during pregnancy or as a consequence of contraceptive treatments. In addition, WD heterozygous carriers occasionally present with abnormally reduced Cp levels. Other considerations for low Cp levels are malabsorption, fulminant hepatic failure or aceruloplasminemia caused by mutations in CP. Therefore, the predictive value of Cp as a unique marker for diagnosis of WD is questionable, principally because many WD patients show bordering levels.
Serum copper
Total serum copper comprises NCC as well as copper contained in Holo-Cp. Total serum copper and Holo-Cp concentrations are positively correlated, and hence, decreased values of Holo-Cp are likely in WD patients. However, higher levels of Holo-Cp are detected for instance, in Wilsonian acute liver failure onset as a result of accumulated hepatic copper release to bloodstream. Moreover, increased NCC is also observed in cases of acute hepatic failure of different aetiology, chronic cholestasis and idiopathic copper toxicosis [13]. High or normal serum copper along with low Cp indicates an increment of serum copper’s NCC fraction, suggestive finding of WD. Copper belonging to the NCC fraction, sometimes not well-named as “free”, becomes harmful when deposited in tissues, mainly in liver and brain. As expected, the results depend on proper Holo-Cp determinations on the same sample. As a result, if Cp levels are determined by immunoassays that include the detection of Apo-Cp and Holo-Cp, NCC estimation for diagnosis and monitoring might be misleading [14].
Urinary copper
In treatment-naïve WD patients, urinary copper excretion (UCE), a reflection of serum NCC, is elevated, and thus, measure of copper in 24-hour urinary collection is a common test for diagnosis of WD. In symptomatic patients, values higher than 1.2 to 2 times the upper limit of normal (ULN, established on 100 µg/24h) are registered, which may indicate possible WD. Urinary copper measurement after stimulation with 1,000 mg D-penicillamine is a test proposed for diagnosis in children, with expected levels >1,600 µg/24h in WD children [15].
Additional blood tests
Typically, persistently elevated liver enzymes, specifically aspartate aminotransferase (AST) and alanine aminotransferase (ALT), is the first disease manifestation that should alert medical specialists of a possible WD case in absence of any known etiology, particularly in young patients. Biochemical parameters, as serum alkaline phosphatase (ALP) and total bilirubin, may be informative at some point to distinguish ALF due to WD from that of different aetiology [13].
Liver biopsy
Clinical practice guidelines only recommend intrahepatic copper quantification if remaining evidences are insufficient to achieve a diagnosis, or if other liver pathology is suspected apart from WD. If other disorders are discarded, a hepatic copper content higher than 250 µg/g dry weight is considered confirmatory of WD. Other studies suggest that this threshold could be fixed in 75 µg/g dry weight for better sensibility [10]. Specific stains (rhodanine or orcein) are only useful in case of copper deposition in lysosomes, a finding rarely present. Since copper deposition across liver structures is dependent of disease stage, quantification of intrahepatic copper is not always reliable. At cirrhosis stage, copper deposition is not homogeneous, so liver sample is usually not representative and copper concentration results are generally underestimated [15].
Early liver histological findings resemble those representatives of non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH), comprising simple steatosis, glycogenated hepatocyte nuclei and focal necrosis. In untreated or late-diagnosed patients with advanced liver disease, copper overload and toxicity lead to major inflammation, fibrosis and eventually cirrhosis.
Neurologic evaluation
Apart from patients with predominant neurological manifestations, clinical evaluation by a neurologist or specialist in movement disorders is recommended in all cases. Two score systems have been developed and validated to help on the assessment of neurological impairment: Unified Wilson’s Disease Rating Scale (UWDRS) and Global Assessment Scale for Wilson’s Disease (GAS for WD). Brain magnetic resonance imaging (MRI) is essential to assess presence of copper deposits and damage on basal ganglia. Common recognized abnormalities include detection of hyperintensities on T2 sequences, mainly in putamen and caudate nucleus; the “face of the giant panda” on the midbrain is considered a characteristic sign. Alterations in brain MRI at diagnosis are present in all neurological patients, between 40-75% of hepatic patients and in 20-30% of asymptomatic individuals [16].
4. A Mendelian disease caused by mutations in ATP7B
WD is a well-established monogenic condition caused by deleterious variants in ATP7B inherited in an autosomal recessive manner [17]. ATP7B exhibits a high allelic heterogeneity. To date, more than 900 variants have been described in ATP7B (HGMD Professional version 2021.1, accessed June 2021). The vast majority are located in the coding and flanking intronic sequences throughout the entire gene, the most frequent being missense and nonsense mutations (>60%).
Stratified genetic analysis is recommended in order to achieve a reasonable diagnostic success rate with the aim to cover the whole ATP7B sequence and the different possible mutations types [18][19]. First, it is advisable to perform the study of the 21 coding exons and flanking intron sequences, since they harbour the majority (~95%) of the clinical variants. This approach can be done by Sanger sequencing, which allows the detection of point mutations as well as small deletions and insertions. In populations in which a small number of pathological changes predominate, the prioritization of these likely founder events is a cost-effective strategy. If no diagnosis is achieved, the possibility of large deletions or insertions must be examined by MLPA (multiplex-ligation probe amplification), in addition to sequencing the promoter. These clinical variants represent ~4% of the total known ATP7B mutations. Finally, the study of the introns must be taken into account if no biallelic mutations have been detected in ATP7B in a patient with clinical suspicion of WD. The diagnostic tools based on NGS (next generation sequencing) allow for studies of large tracts, and hence, the analysis of the entire 75 kb sequence of ATP7B (exons, introns, 3’ and 5’-UTR regions, and promoter) [18]. In fact, using a custom NGS tool, one deep intronic deleterious mutation was published located in intron 12, c.2866-1521G>A [20]. Of note, NGS tools also allow the identification of large CNVs (copy number variants).
The widely accepted prevalence of WD was estimated in 1984 by Scheinberg and Sternlieb [21]: 1:30,000 (= 3.3/100,000). The frequency of carriers, calculated according to Hardy-Weinberg equilibrium and full penetrance, is considered to be 1:90 [22]. Of course, prevalence increases in closed/isolated populations, as is the case of the islands of Canary (Spain; 8.1/100,000), Sardinia (Italy; 37/100,000), or Kalymmos (Greece; 135/100,000) [23] [24]. Many studies have intended to establish accurate values of prevalence with findings related to the strategy applied such that higer prevalence rates have been described in mutational screening studies compared to those based on public health registries. These discrepancies are mainly due to differences between genetic and clinical prevalences. As aforementioned, diagnosis of WD is based on the Leipzig score and relies on imperfect clinical, biochemical, histological, and genetic findings. Despite technological advances, only one mutation or no mutation is identified in a relevant proportion of patients (~1-27%) [19]. Gao et al. [24] established a clinical prevalence of 1.38 and a genetic prevalence of 12.7 per 100,000 in a meta-analysis of previous epidemiological and genetic studies, respectively. As a whole, genetic prevalence is much higher and highlights that WD is an underdiagnosed disease.
5. Wilson-like: genetic diseases that mimic the Wilson phenotype
WD is clearly established as a monogenic disorder caused by ATP7B mutations. However, disparate inherited diseases that mimic a Wilson-like phenotype are known.
Congenital disorders of glycosylation (CDG) are a heterogeneous group affecting multiple glycosylation pathways and they can display phenotypic similarities with WD [25][18]. Many CDGs show multisystemic clinical outcomes. Patients with CDG linked to CCDC115 mutations (MIM 616828) present hepatosplenomegaly, neurological affectations, elevated serum ALT and ALP levels, mild hypercholesterolemia, low serum Cp and Wilson-like copper disturbances. On the other hand, individuals with TMEM199 mutations exhibit steatosis, elevated serum levels of ALT and ALP, hypercholesterolemia, low serum Cp and slightly alterations of copper metabolism; they do not develop hepatosplenomegaly or neuropsychiatric disturbances [26].
Progressive familial intrahepatic cholestasis (PFIC) is a heterogenous group of autosomal recessive liver disorders characterized by defective bile transport. One of its early onset forms, PFIC3 (MIM 602347), is caused by mutations in the ABCB4 gene which results in impaired phospholipid secretion, triggering cholestasis, hepatic fibrosis, cirrhosis and hepatocellular failure [27].
Aceruloplasminaemia (MIM 604290) is an autosomal recessive disorder caused by mutations in the CP gene, which encodes ceruloplasmin. Neuropsychiatric manifestations may include dementia, cognitive deficit, behaviour alterations, tremor, chorea, cerebellar ataxia, nystagmus, dysarthria, rigidity and akinesia [28]. Normal concentration of hepatic copper and in urinary copper excretion can be discriminant indicators between WD and aceruloplasminaemia.
Menkes disease (MNK; MIM 309400) is an X-linked syndrome caused by mutations in ATP7A. ATP7A, as well as ATP7B, is a transmembrane copper transporter and its dysfunction turns into deficient copper transfer to secretory pathway. Allelic disorders of MNK disease are occipital horn syndrome (OHS; MIM 3040150) and X-linked distal spinal muscular atrophy 3 (SMAX3; MIM 300489). These three entities are best described as a clinical continuum spectrum from severe to mild forms. MNK is characterized by epilepsy, progressive neurodegeneration, conjunctive tissue abnormalities, a distinctive hair appearance and short lifespan [29].
MEDNIK syndrome (Mental retardation, Enteropathy, Deafness, Neuropathy, Ichthyosis and Keratoderma; MIM 609313) is a rare neurocutaneous disorder with multisystem involvement, caused by AP1S1 mutations and inherited in an autosomal recessive fashion. Patients display a perplexing picture of MNK disease and WD. Some neurological, skeletal and cutaneous signs, as well as low copper and Cp levels in plasma, resemble MNK but in a milder form. On the other hand, hepatic damage, increased urinary copper excretion and bilateral T2 hyperintensity of basal ganglia (caudate and putamen) are typical clinical manifestations of WD [30].
Alagille syndrome (ALGS; MIM 118450) is a multisystem disorder caused by mutations in JAG1 or NOTCH2 genes transmitted in an autosomal dominant way. ALGS is characterized by the scarcity of bile ducts in liver biopsy and, at least, the presence of three of five clinical features: cholestasis, congenital heart defects, ocular abnormalities, bone anomalies and characteristic facies. Renal disease, development and growth retardation may occur less frequently [31].
Idiopathic copper toxicosis (ICP) is referred to a type of infancy and childhood liver disease with abnormal copper accumulation whose etiology is poorly understood. Symptoms can include hepatosplenomegaly, marked fibrosis and abdominal distention, occasionally accompanied by ascites, lethargy, malaise, fever, anaemia and, rarely, recurrent jaundice. Serum copper and ceruloplasmin concentrations are normal or slightly elevated [32].
6. Conclusions
Diagnosis of WD should be suspected when faced with patients, particularly children, adolescent or young adults with any manifestations of liver disease without known etiological factor and/or when signs of neurological disease are also present. Nowadays, there are multiple diagnostic and treatment options for patients suffering from this disorder. While many patients are diagnosed and adequately assessed with current available methods, diagnosis has revealed to be more challenging in other patients stuck in a diagnostic uncertainty characterized by bordering ceruloplasmin levels, inconclusive genetic findings and unclear clinical phenotypes. Patient prognosis is based on early diagnosis and adequate long-life therapy. Delay in diagnosis, and therefore, in starting decoppering treatment can result in disastrous consequences. Technological advances in genetics tests, neuroimaging, characterization of biomarkers, and advanced therapies may provide confidence for a more accurate detection and management of WD in coming years.
References
- M.D. S. A. Kinnier Wilson; PROGRESSIVE LENTICULAR DEGENERATION: A FAMILIAL NERVOUS DISEASE ASSOCIATED WITH CIRRHOSIS OF THE LIVER. Brain 1912, 34, 295-507, 10.1093/brain/34.4.295.
- Alfred Rumpel; Über das Wesen und die Bedeutung der Leberveränderungen und der Pigmentierungen bei den damit verbundenen Fällen von Pseudosklerose, zugleich ein Beitrag zur Lehre von der Pseudosklerose (Westphal-Strümpell). Journal of Neurology 1913, 49, 54-73, 10.1007/bf01760543.
- J. N. Cumings; THE COPPER AND IRON CONTENT OF BRAIN AND LIVER IN THE NORMAL AND IN HEPATO-LENTICULAR DEGENERATION. Brain 1948, 71, 410-415, 10.1093/brain/71.4.410.
- Aurélia Poujois; France Woimant; Wilson's disease: A 2017 update. Clinics and Research in Hepatology and Gastroenterology 2018, 42, 512-520, 10.1016/j.clinre.2018.03.007.
- Karl Heinz Weiss; Daniel Nils Gotthardt; Daniela Klemm; Uta Merle; Daniela Ferenci–Foerster; Mark Schaefer; Peter Ferenci; Wolfgang Stremmel; Zinc Monotherapy Is Not as Effective as Chelating Agents in Treatment of Wilson Disease. Gastroenterology 2011, 140, 1189-1198.e1, 10.1053/j.gastro.2010.12.034.
- Karl Heinz Weiss; Frederick K Askari; Anna Czlonkowska; Peter Ferenci; Jeff M Bronstein; Danny Bega; Aftab Ala; David Nicholl; Susan Flint; Lars Olsson; et al.Thomas PlitzCarl BjartmarMichael L Schilsky Bis-choline tetrathiomolybdate in patients with Wilson's disease: an open-label, multicentre, phase 2 study. The Lancet Gastroenterology & Hepatology 2017, 2, 869-876, 10.1016/s2468-1253(17)30293-5.
- Oihana Murillo; Daniel Moreno; Cristina Gazquez; Miren Barberia; Itziar Cenzano; Iñigo Navarro-Blasco; Iker Uriarte; Victor Sebastian; Manuel Arruebo; Veronica Ferrer; et al.Bernard BénichouJean Philippe CombalJesus PrietoRuben Hernandez‐AlcocebaGloria Gonzalez Aseguinolaza Liver Expression of a MiniATP7B Gene Results in Long‐Term Restoration of Copper Homeostasis in a Wilson Disease Model in Mice. Hepatology 2019, 70, 108-126, 10.1002/hep.30535.
- Samuel Shribman; Thomas Warner; James S. Dooley; Clinical presentations of Wilson disease.. Annals of Translational Medicine 2019, 7, S60-S60, 10.21037/atm.2019.04.27.
- Anna Członkowska; Tomasz Litwin; Petr Dusek; Peter Ferenci; Svetlana Lutsenko; Valentina Medici; Janusz K. Rybakowski; Karl Heinz Weiss; Michael L. Schilsky; Wilson disease. Nature Reviews Disease Primers 2018, 4, 1-20, 10.1038/s41572-018-0018-3.
- Richard Rosencrantz; Michael Schilsky; Wilson Disease: Pathogenesis and Clinical Considerations in Diagnosis and Treatment. Seminars in Liver Disease 2011, 31, 245-259, 10.1055/s-0031-1286056.
- European Association for the Study of the Liver; EASL Clinical Practice Guidelines: Wilson’s disease. Journal of Hepatology 2012, 56, 671-685, 10.1016/j.jhep.2011.11.007.
- Ana Sánchez-Monteagudo; Edna Ripollés; Marina Berenguer; Carmen Espinós; Wilson’s Disease: Facing the Challenge of Diagnosing a Rare Disease. Biomedicines 2021, 9, 1100, 10.3390/biomedicines9091100.
- Chloe M. Mak; Biochemical Diagnosis of Wilson Disease. Clinical and Translational Perspectives on WILSON DISEASE 2018, 1, 237-248, 10.1016/b978-0-12-810532-0.00021-5.
- Isabelle Mohr; Karl Heinz Weiss; Biochemical markers for the diagnosis and monitoring of Wilson Disease. Clinical Biochemist Reviews 2019, 40, 59-77, 10.33176/aacb-18-00014.
- Eve A. Roberts; Michael L. Schilsky; Diagnosis and treatment of Wilson disease: An update. Hepatology 2008, 47, 2089-2111, 10.1002/hep.22261.
- Anna Członkowska; Tomasz Litwin; Grzegorz Chabik; Wilson disease. Handbook of Clinical Neurology 2017, 142, 101-119, 10.1016/b978-0-444-63625-6.00010-0.
- R. E. Tanzi; K. Petrukhin; Igor Chernov; Jean-Luc Pellequer; W. Wasco; B. Ross; D. M. Romano; Enrico Parano; L. Pavone; L. M. Brzustowicz; et al.Marcella DevotoJ. PeppercornAshley BushI. SternliebM. PirastuJ. F. GusellaO. EvgrafovG. K. PenchaszadehB. HonigI. S. EdelmanM. B. SoaresI. H. ScheinbergT. C. Gilliam The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nature Genetics 1993, 5, 344-350, 10.1038/ng1293-344.
- Ana Sánchez-Monteagudo; María Álvarez-Sauco; Isabel Sastre; Irene Martínez-Torres; Vincenzo Lupo; Marina Berenguer; Carmen Espinós; Genetics of Wilson disease and Wilson‐like phenotype in a clinical series from eastern Spain. Clinical Genetics 2020, 97, 758-763, 10.1111/cge.13719.
- Carmen Espinós; Peter Ferenci; Are the new genetic tools for diagnosis of Wilson disease helpful in clinical practice?. JHEP Reports 2020, 2, 100114, 10.1016/j.jhepr.2020.100114.
- France Woimant; Aurelia Poujois; Adrien Bloch; Tabaras Jordi; Jean‐Louis Laplanche; Hélène Morel; Corinne Collet; A novel deep intronic variant in ATP7B in five unrelated families affected by Wilson disease. Molecular Genetics & Genomic Medicine 2020, 8, 1-9, 10.1002/mgg3.1428.
- H. Scheinberg, I. Sternlieb; Wilson's disease (a volume in the major problems in internal medicine series) . Annals of Neurology 1984, 16, 626-626, 10.1002/ana.410160531.
- Thomas Damgaard Sandahl; Tea Lund Laursen; Ditte Emilie Munk; Hendrik Vilstrup; Karl Heinz Weiss; Peter Ott; The Prevalence of Wilson’s Disease: An Update. Hepatology 2019, 71, 722-732, 10.1002/hep.30911.
- Luis García-Villarreal; Susan Daniels; Sarah H. Shaw; David Cotton; Margaret Galvin; Jeanne Geskes; Paula Bauer; Angel Sierra-Hernández; Alan Buckler; Antonio Tugores; et al. High prevalence of the very rare wilson disease gene mutation Leu708Pro in the Island of Gran Canaria (Canary Islands, Spain): A genetic and clinical study. Hepatology 2000, 32, 1329-1336, 10.1053/jhep.2000.20152.
- Jiali Gao; Simon Brackley; Jake P. Mann; The global prevalence of Wilson disease from next-generation sequencing data. Genetics in Medicine 2018, 21, 1155-1163, 10.1038/s41436-018-0309-9.
- Muriel Girard; Aurélia Poujois; Monique Fabre; Florence Lacaille; Dominique Debray; Marlène Rio; François Fenaille; Sophie Cholet; Coralie Ruel; Elizabeth Caussé; et al.Janick SelvesLaure Bridoux-HennoFrance WoimantThierry DupréSandrine Vuillaumier-BarrotNathalie SetaLaurent AlricPascale de LonlayArnaud Bruneel CCDC115-CDG: A new rare and misleading inherited cause of liver disease. Molecular Genetics and Metabolism 2018, 124, 228-235, 10.1016/j.ymgme.2018.05.002.
- Jos C. Jansen; Sharita Timal; Monique van Scherpenzeel; Helen Michelakakis; Dorothée Vicogne; Angel Ashikov; Marina Moraitou; Alexander Hoischen; Karin Huijben; Gerry Steenbergen; et al.Marjolein A.W. Van Den BoogertFrancesco PortaPier Luigi CalvoMersyni MavrikouGiovanna CenacchiGeert Van Den BogaartJody SalomonA.G. (Onno) HolleboomRichard RodenburgJoost P.H. DrenthMartijn HuynenRon A. WeversEva MoravaFrançois FoulquierJoris A. VeltmanDirk J. Lefeber TMEM199 Deficiency Is a Disorder of Golgi Homeostasis Characterized by Elevated Aminotransferases, Alkaline Phosphatase, and Cholesterol and Abnormal Glycosylation. The American Journal of Human Genetics 2016, 98, 322-330, 10.1016/j.ajhg.2015.12.011.
- Sriram Amirneni; Nils Haep; Mohammad A Gad; Alejandro Soto-Gutierrez; James E Squires; Rodrigo M Florentino; Molecular overview of progressive familial intrahepatic cholestasis. World Journal of Gastroenterology 2020, 26, 7470-7484, 10.3748/wjg.v26.i47.7470.
- Mária Ondrejkovičová; Sylvia Dražilová; Monika Drakulová; Juan López Siles; Renáta Zemjarová Mezenská; Petra Jungová; Martin Fabián; Boris Rychlý; Miroslav Žigrai; New mutation of the ceruloplasmin gene in the case of a neurologically asymptomatic patient with microcytic anaemia, obesity and supposed Wilson’s disease. BMC Gastroenterology 2020, 20, 1-6, 10.1186/s12876-020-01237-8.
- Nina Horn; Pernilla Wittung-Stafshede; ATP7A-Regulated Enzyme Metalation and Trafficking in the Menkes Disease Puzzle. Biomedicines 2021, 9, 391, 10.3390/biomedicines9040391.
- Diego Martinelli; Lorena Travaglini; Christian A. Drouin; Irene Ceballos-Picot; Teresa Rizza; Enrico Bertini; Rosalba Carrozzo; Stefania Petrini; Pascale De Lonlay; Maya El Hachem; et al.Laurence HubertAlexandre MontpetitGiuliano TorreCarlo Dionisi-Vici MEDNIK syndrome: a novel defect of copper metabolism treatable by zinc acetate therapy. Brain 2013, 136, 872-881, 10.1093/brain/awt012.
- Melissa A. Gilbert; Kathleen M. Loomes; Alagille syndrome and non-syndromic paucity of the intrahepatic bile ducts. Translational Gastroenterology and Hepatology 2021, 6, 22-22, 10.21037/tgh-2020-03.
- Alicia A. Taylor; Joyce S. Tsuji; Michael R. Garry; Margaret E. McArdle; William L. Goodfellow; William J. Adams; Charles A. Menzie; Critical Review of Exposure and Effects: Implications for Setting Regulatory Health Criteria for Ingested Copper. Environmental Management 2019, 65, 131-159, 10.1007/s00267-019-01234-y.


