 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Declan McCole | + 1745 word(s) | 1745 | 2021-09-02 08:44:49 | | | |
| 2 | Dean Liu | Meta information modification | 1745 | 2021-09-03 08:10:30 | | |
Video Upload Options
The intestinal epithelial barrier consists of multiple elements that contribute to its function as a physical, chemical, and immunological defense. While the mucus layer, intestinal epithelium, and the underlying immune cells in the lamina propria comprise a major component of the defense response, intestinal epithelial permeability is regulated by tight junctions.
1. Introduction
Inflammatory bowel diseases (IBD) are chronic inflammatory disorders of the gastrointestinal tract and primarily include Crohn’s disease (CD) and ulcerative colitis (UC). Inflammation in CD predominantly occurs in the small intestine while UC is commonly associated with inflammation of the rectum and areas of the colon [1][2][3]. Being a transmural disease, the presence of granulomas, focal crypt architectural abnormalities, and mucin preservation at active sites are frequently seen in CD patients [4]. However, focal or diffuse basal plasmacytosis, widespread mucosal or crypt architectural distortion, and mucin depletion are commonly seen in UC patients [4][5][6]. Collectively, patients with IBD can present a multitude of symptoms including diarrhea, abdominal pain, weight loss and in severe cases, nutrient deficiency, and anemia [4]. Furthermore, patients with IBD are at risk of developing complications including intestinal obstruction and colorectal cancer that can result in hospitalization and subsequent surgical interventions [4]. Despite the abundance of therapeutic strategies for IBD, many patients fail to respond appropriately to the available treatments [1][3]. Moreover, the prevalence of IBD is consistently rising with approximately 7 million cases globally [7].
Inflammatory bowel diseases are multifactorial disorders that manifest from an abnormal immune response towards luminal contents in the digestive tract in processes driven by genetic and environmental risk factors [8][9]. Currently, over two hundred SNPs have been linked to the pathogenesis of IBD, many of which are involved in regulating intestinal epithelial barrier function [8]. Disruption of the intestinal epithelial barrier, via increased intestinal permeability, is a feature of many chronic inflammatory disorders including IBD. However, it was uncertain whether the increased intestinal permeability found in patients with CD or UC is an associated finding or if it plays a significant role in disease pathogenesis [10].
Intriguingly, a prospective study by Turpin et al. demonstrated that increased intestinal permeability is associated with the later development of CD [11]. This finding has substantial implications in understanding the pathogenesis of IBD as it provides the most robust evidence of increased intestinal permeability predisposing to subsequent onset of CD. Variants in genes belonging to the Janus kinase (JAK)—signal transducer and activator of transcription (STAT) signaling pathway, as well as members of the protein tyrosine phosphatase (PTP) family that act as negative regulators of JAK-STATs, are associated with increased IBD risk [8][9]. JAK-STAT proteins are responsible for mediating receptor signaling of numerous IBD-associated cytokines involved in regulating intestinal permeability [9]. With regulation of intestinal permeability being an essential component of proper intestinal epithelial barrier function, developing therapeutics to target signaling cascades such as the JAK-STAT pathway represent an attractive therapeutic avenue for patients with IBD.
2. Intestinal Permeability in IBD
The intestinal epithelial barrier is a single layer of epithelial cells that is essential for regulation of intestinal homeostasis and mediates communication between the intestinal microflora and the immune system [12][13]. In addition to being a dynamic interface between the luminal contents and the host immune system, the epithelium is the first site of exposure to many of the environmental factors that can trigger disease activity in chronic IBD [12][13]. With the epithelium neighboring the luminal microbes and the underlying immune system, the integrity of the epithelial lining is critical to avoid excessive contact of pathogenic antigens with lamina propria immune cells. Aside from serving as a physical barrier, intestinal epithelial cells can also endocytose bacteria, sequester and neutralize toxins, and detect pathogen-associated molecular patterns (PAMPs) [14]. Furthermore, the epithelium can respond to intestinal damage by secreting factors that contribute to epithelial restitution, initiating wound repair, and activating the underlying innate and adaptive immunity [13]. However, disruption of this selective barrier can lead to chronic intestinal inflammation through the uncontrolled uptake of foreign antigens and the overstimulation of the mucosal immune system [15]. Along with its innate immune functions, the intestinal epithelium can perform a variety of specialized tasks due to its ability to form a tightly regulated and selectively permeable barrier. This leaky barrier is essential for the epithelium to generate ion solute concentration gradients to appropriately absorb nutrients and water [13]. Moreover, a regulated intestinal barrier is required for controlled antigen transport to the resident immune cells in the gut-associated lymphoid tissue, thus supporting the maturation of the immune system [16]. Therefore, proper function of the intestinal epithelial barrier is essential for prevention of intraluminal pathogen invasion, regulated intake of essential nutrients, and managing the intestinal immune response.
These clinical findings align with studies in animal models of IBD demonstrating that increased intestinal paracellular permeability precedes the onset of inflammation in the gut. The SAMP1/YitFc mouse model of chronic ileitis was described to have a permeability defect before the presence of ileal inflammation [17]. SAMP mice exhibited profound epithelial barrier alterations with reduced transepithelial electrical resistance (TEER) and elevated LMR [17]. Similarly, mdr1a −/− mice, lacking the multidrug resistance gene that encodes the p-glycoprotein transporter, presented with chronic inflammation of the gut when exposed to a “normal” microbiota [18]. A defect in mdr1a is likely to predispose the intestinal epithelium to bacterial invasion and infection [18]. Madsen et al., showed that increased intestinal permeability is evident before intestinal inflammation in Il10 −/− mice [19]. Of interest, treatment of Il10 −/− mice with the probiotic compound, VSL#3, results in the normalization of colonic epithelial barrier along with a reduction in levels of pro-inflammatory cytokines including TNF-α and IFN-γ [19].
Mechanistically, alterations in expression of tight junction proteins have been reported to affect intestinal permeability and contribute to the pathogenesis of IBD. Mice with intestinal deletion of claudin-7 exhibited colonic inflammation even though tight junction structures were intact, suggesting differences in expression of certain tight junction proteins can increase or reduce intestinal paracellular permeability [20]. Similar effects were also seen in colonic tissues from patients with active CD. These patients showed impaired intestinal barrier function as indicated by a dramatic reduction in TEER [21]. Moreover, this drop in intestinal barrier function was associated with a decrease in tight junction strand number and continuity, in addition to an increase in epithelial apoptosis [21]. The size of the discontinuity of these tight junction strand breaks were considered large enough to facilitate paracellular passage of foreign antigens [21]. Expression of the barrier sealing tight junction proteins, occluding, and claudins -3, -5, and -8, were also dramatically decreased in patients with active CD while increased expression of the pore-forming claudin-2 was upregulated [21].
Consistent with the clinical findings of Turpin et al., animal studies also indicate that increased intestinal paracellular permeability by itself is insufficient to cause colitis [11]. Su et al. , demonstrated that constitutively-active myosin light chain kinase transgenic mice had significant barrier loss but grew similarly to their gender-matched littermates and did not develop intestinal inflammation [22]. However, when colitis was induced in these mice, they suffered from more severe inflammation and had significantly shorter survival times [22]. Furthermore, junctional adhesion molecule-A (JAM-A) deficient mice had normal intestinal epithelial architecture in their colonic mucosa despite the increased presence of lymphoid aggregates and reduced intestinal barrier function [23]. Intriguingly, these JAM-A deficient mice were more sensitive to dextran sulfate sodium (DSS)-induced colitis [23][24]. Overall, these studies support the emerging hypothesis that an impaired barrier defect along with a trigger of an inappropriate immune response is critical for intestinal inflammation. Nevertheless, maintaining intestinal barrier function via regulating tight junction proteins is necessary for preventing intestinal inflammation ( Figure 1 ) and developing therapeutics that target their regulators may be a viable clinical approach in patients with IBD.
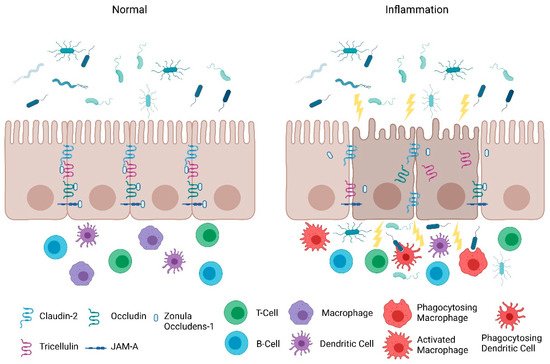
3. Protection of the Intestinal Epithelial Barrier
The intestinal epithelial barrier consists of multiple elements that contribute to its function as a physical, chemical, and immunological defense. While the mucus layer, intestinal epithelium, and the underlying immune cells in the lamina propria comprise a major component of the defense response, intestinal epithelial permeability is regulated by tight junctions. In the following section, we will discuss the role and mechanisms of tight junctions as a selectively permeable barrier, and the significance of the JAK-STAT pathway in barrier permeability.
4. JAK-STAT Signaling in Apoptosis and Necroptosis of Intestinal Epithelial Cells
Along with its vital role in regulating intestinal permeability via tight junction proteins, epithelial barrier function can be altered via the “unrestricted pathway”, which is primarily associated with cell loss due to death or shedding [25]. This can also be modulated by JAK-STATs as JAK-STAT signaling can mediate different forms of cell death, apoptosis, and necroptosis in IECs. Deletion of Stat1 in mouse ileal IECs protected against IFN-λ-induced necroptosis and apoptosis in Paneth cells [26]. When treated with tofacitinib, IFN-λ-treated small intestinal organoids had lower gene transcription of the cell death mediators, Mlkl and Caspase - 8 [26]. Notably, the addition of tofacitinib restored the viability of the organoids and prevented Paneth cell death in response to IFN-λ [26]. Filgotinib, a novel selective JAK1 inhibitor, showed similar effects and interfered with STAT1 phosphorylation [26].
In a related study, deletion of Stat1 in mouse ileal IECs lacking Caspase - 8 partially recovered Paneth cell numbers and was associated with reduced Mlkl expression and a lower number of TUNEL (terminal deoxynucleotide transferase dUTP nick end labeling) positive cells [27] . In contrast, Paneth cell function did not return to normal levels, indicating an alternative mechanism that regulates Paneth cell activity [27]. Surprisingly, Stat1 was demonstrated to be unessential in regulating cell death in the colon when inflammation was induced with DSS [27].
Of interest, an increase of reserve stem cells was observed after treatment of enteroids and mice with TNF-α and IFN-γ, suggesting cytokines released during mucosal inflammation are required for the regenerative response [28]. Notably, the JAK-STAT1 pathway was found to be necessary for activation of reserve stem cells during inflammation as pre-treatment of enteroid cultures with tofacitinib prevented the IFN-γ-induced increase in reserve stem cell number [28]. These findings demonstrate an alternative role of the JAK-STAT pathway in facilitating the restitution of the epithelial barrier.
References
- Lichtenstein, G.R.; Loftus, E.; Isaacs, K.L.; Regueiro, M.D.; Gerson, L.B.; Sands, B.E. ACG Clinical Guideline: Management of Crohn’s Disease in Adults. Am. J. Gastroenterol. 2018, 113, 481–517.
- Torres, J.; Bonovas, S.; Doherty, G.; Kucharzik, T.; Gisbert, J.P.; Raine, T.; Adamina, M.; Armuzzi, A.; Bachmann, O.; Bager, P.; et al. ECCO Guidelines on Therapeutics in Crohn’s Disease: Medical Treatment. J. Crohn’s Colitis 2019, 14, 4–22.
- Rubin, D.T.; Ananthakrishnan, A.N.; Siegel, C.A.; Sauer, B.G.; Long, M.D. ACG Clinical Guideline: Ulcerative Colitis in Adults. Am. J. Gastroenterol. 2019, 114, 384–413.
- Maaser, C.; Sturm, A.; Vavricka, S.R.; Kucharzik, T.; Fiorino, G.; Annese, V.; Calabrese, E.; Baumgart, D.C.; Bettenworth, D.; Borralho Nunes, P.; et al. ECCO-ESGAR guideline for diagnostic assessment in IBD part 1: Initial diagnosis, monitoring of known IBD, detection of complications. J. Crohn’s Colitis 2019, 13, 144–164.
- Langner, C.; Magro, F.; Driessen, A.; Ensari, A.; Mantzaris, G.J.; Villanacci, V.; Becheanu, G.; Nunes, P.B.; Cathomas, G.; Fries, W.; et al. The histopathological approach to inflammatory bowel disease: A practice guide. Virchows Arch. 2014, 464, 511–527.
- Magro, F.; Langner, C.; Driessen, A.; Ensari, A.; Geboes, K.; Mantzaris, G.; Villanacci, V.; Becheanu, G.; Nunes, P.B.; Cathomas, G.; et al. European consensus on the histopathology of inflammatory bowel disease. J. Crohn’s Colitis 2013, 7, 827–851.
- Alatab, S.; Sepanlou, S.G.; Ikuta, K.; Vahedi, H.; Bisignano, C.; Safiri, S.; Sadeghi, A.; Nixon, M.R.; Abdoli, A.; Abolhassani, H.; et al. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30.
- Spalinger, M.R.; Sayoc-Becerra, A.; Ordookhanian, C.; Canale, V.; Santos, A.N.; King, S.J.; Krishnan, M.; Nair, M.G.; Scharl, M.; McCole, D.F. The JAK Inhibitor Tofacitinib Rescues Intestinal Barrier Defects Caused by Disrupted Epithelial-macrophage Interactions. J. Crohn’s Colitis 2021, 15, 471–484.
- Sayoc-Becerra, A.; Krishnan, M.; Fan, S.; Jimenez, J.; Hernandez, R.; Gibson, K.; Preciado, R.; Butt, G.; McCole, D.F. The JAK-Inhibitor Tofacitinib Rescues Human Intestinal Epithelial Cells and Colonoids from Cytokine-Induced Barrier Dysfunction. Inflamm. Bowel Dis. 2020, 26, 407–422.
- Michielan, A.; D’Incà, R. Intestinal Permeability in Inflammatory Bowel Disease: Pathogenesis, Clinical Evaluation, and Therapy of Leaky Gut. Mediat. Inflamm. 2015, 2015, 628157.
- Turpin, W.; Lee, S.H.; Raygoza Garay, J.A.; Madsen, K.L.; Meddings, J.B.; Bedrani, L.; Power, N.; Espin-Garcia, O.; Xu, W.; Smith, M.I.; et al. Increased Intestinal Permeability Is Associated with Later Development of Crohn’s Disease. Gastroenterology 2020, 159, 2092–2100.e5.
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 9–21.
- Chelakkot, C.; Ghim, J.; Ryu, S.H. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp. Mol. Med. 2018, 50, 1–9.
- Neal, M.D.; Leaphart, C.; Levy, R.; Prince, J.; Billiar, T.R.; Watkins, S.; Li, J.; Cetin, S.; Ford, H.; Schreiber, A.; et al. Enterocyte TLR4 Mediates Phagocytosis and Translocation of Bacteria Across the Intestinal Barrier. J. Immunol. 2006, 176, 3070–3079.
- McCole, D.F. IBD Candidate Genes and Intestinal Barrier Regulation. Inflamm. Bowel Dis. 2014, 20, 1829–1849.
- Martini, E.; Krug, S.; Siegmund, B.; Neurath, M.F.; Becker, C. Mend Your Fences. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 33–46.
- Olson, T.S.; Reuter, B.K.; Scott, K.G.-E.; Morris, M.A.; Wang, X.-M.; Hancock, L.N.; Burcin, T.L.; Cohn, S.M.; Ernst, P.; Cominelli, F.; et al. The primary defect in experimental ileitis originates from a nonhematopoietic source. J. Exp. Med. 2006, 203, 541–552.
- Resta-Lenert, S.; Smitham, J.; Barrett, K.E. Epithelial dysfunction associated with the development of colitis in conventionally housed mdr1a−/− mice. Am. J. Physiol. Liver Physiol. 2005, 289, G153–G162.
- Madsen, K.; Cornish, A.; Soper, P.; McKaigney, C.; Jijon, H.; Yachimec, C.; Doyle, J.; Jewell, L.; De Simone, C. Probiotic bacteria enhance murine and human intestinal epithelial barrier function. Gastroenterology 2001, 121, 580–591.
- Tanaka, H.; Takechi, M.; Kiyonari, H.; Shioi, G.; Tamura, A.; Tsukita, S. Intestinal deletion of Claudin-7 enhances paracellular organic solute flux and initiates colonic inflammation in mice. Gut 2015, 64, 1529–1538.
- Zeissig, S.; Bürgel, N.; Günzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72.
- Su, L.; Shen, L.; Clayburgh, D.; Nalle, S.C.; Sullivan, E.A.; Meddings, J.B.; Abraham, C.; Turner, J.R. Targeted Epithelial Tight Junction Dysfunction Causes Immune Activation and Contributes to Development of Experimental Colitis. Gastroenterology 2009, 136, 551–563.
- Laukoetter, M.G.; Nava, P.; Lee, W.Y.; Severson, E.A.; Capaldo, C.T.; Babbin, B.A.; Williams, I.R.; Koval, M.; Peatman, E.; Campbell, J.A.; et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J. Exp. Med. 2007, 204, 3067–3076.
- Vetrano, S.; Rescigno, M.; Cera, M.R.; Correale, C.; Rumio, C.; Doni, A.; Fantini, M.; Sturm, A.; Borroni, E.; Repici, A.; et al. Unique Role of Junctional Adhesion Molecule-A in Maintaining Mucosal Homeostasis in Inflammatory Bowel Disease. Gastroenterology 2008, 135, 173–184.
- Marchiando, A.M.; Shen, L.; Graham, W.; Edelblum, K.L.; Duckworth, C.; Guan, Y.; Montrose, M.H.; Turner, J.R.; Watson, A.J. The Epithelial Barrier Is Maintained by In Vivo Tight Junction Expansion during Pathologic Intestinal Epithelial Shedding. Gastroenterology 2011, 140, 1208–1218.e2.
- Günther, C.; Ruder, B.; Stolzer, I.; Dorner, H.; He, G.-W.; Chiriac, M.T.; Aden, K.; Strigli, A.; Bittel, M.; Zeissig, S.; et al. Interferon Lambda Promotes Paneth Cell Death Via STAT1 Signaling in Mice and Is Increased in Inflamed Ileal Tissues of Patients with Crohn’s Disease. Gastroenterology 2019, 157, 1310–1322.e13.
- Stolzer, I.; Dressel, A.; Chiriac, M.T.; Neurath, M.F.; Günther, C. An IFN-STAT Axis Augments Tissue Damage and Inflammation in a Mouse Model of Crohn’s Disease. Front. Med. 2021, 8, 667.
- Richmond, C.A.; Rickner, H.; Shah, M.S.; Ediger, T.; Deary, L.; Zhou, F.; Tovaglieri, A.; Carlone, D.L.; Breault, D.T. JAK/STAT-1 Signaling Is Required for Reserve Intestinal Stem Cell Activation during Intestinal Regeneration Following Acute Inflammation. Stem Cell Rep. 2018, 10, 17–26.


