 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | alain couvineau | + 1912 word(s) | 1912 | 2021-08-20 14:39:42 | | | |
| 2 | Conner Chen | Meta information modification | 1912 | 2021-09-03 09:50:50 | | |
Video Upload Options
Orexin neuropeptides (Orexin-A/hypocretin-1 and Orexin-B/hypocretins-2) produced from the same precursor in hypothalamus. These two neurotransmitters and their receptors (OX1R and OX1R), present in the central and peripheral nervous system, play a major role in wakefulness but also in drug addiction, food consumption, homeostasis, hormone secretion, reproductive function, lipolysis and blood pressure regulation. With respect to these biological functions, orexins were involved in various pathologies encompassing narcolepsy, neurodegenerative diseases, chronic inflammations, metabolic syndrome and cancers. The expression of OX1R in various cancers including colon, pancreas and prostate cancers associated with its ability to induce a proapoptotic activity in tumor cells, suggested that the orexins/OX1R system could have a promising therapeutic role.
1. Introduction
The identification of orexin, also termed hypocretin, is still young in science history [1]. It was in the late 1990s, when two independent groups identified and characterized these neuropeptides in mouse hypothalamus [2][3]. Orexins (for more clarity in this text, the single term “orexin” will be retained) were encoded by the same gene (hcrt) constituted of two exons producing a unique precursor named prepro-orexin which provided two peptide isoforms, orexin-A (OxA also termed hypocretin-1) and orexin-B (OxB also termed hypocretin-2) [4]. The major role of orexins in the central nervous system (CNS) is to regulate wakefulness [5]. The dysregulation of orexin production in hypothalamus as well as the loss of orexins neurons leads to narcolepsy associated with cataplexy designated as narcolepsy type I [5]. [In addition to sleep regulation, orexins control energy homeostasis, reward seeking, food consumption, drug addiction and motivation [6][7]. Although the central action of orexins has been widely described, some studies have demonstrated that orexins also play a physiological role in the peripheral nervous system (6). Orexins are able to regulate reproductive and neuroendocrine functions, gastrointestinal motility, blood pressure, metabolism and energy balance. However, few reports have been dedicated to orexins’ role in the peripheral nervous system, indicating that these peripheral actions remain relatively controversial [6]. Orexins mediate these biological actions by activating two orexin receptor subtypes that have been identified as orexin receptor type 1 (OX1R) and orexin receptor type 2 (OX2R). These two receptors are associated with Gq protein and belong to the G protein-coupled receptor (GPCR) family [8]. The activation of these receptors by orexins leads to the production of intracellular Ca2+, involving the activation of αq, the phospholipase C and inositol triphosphate (IP3) production (Figure 1) [8].
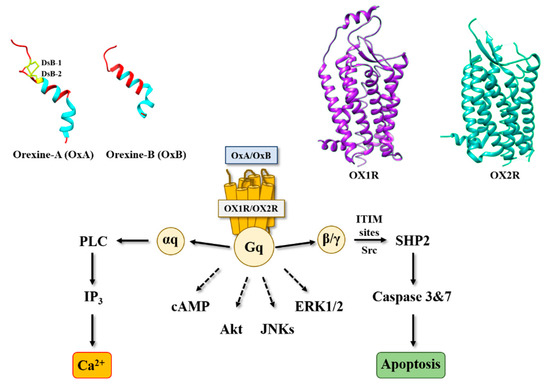
Figure 1. Structure of orexins and their receptors and main signaling pathways activated by orexins in cancer cells. DsB, disulfide bridge; IP3, inositol tri-phosphate; PLC, phospholipase; SHP2, Src homology 2 (SH2) domains of SH2-containing phosphatase 2; cAMP, cyclic adenosine monophosphate; Akt, protein kinase B; JNKs, c-jun N-terminal kinases; ERK1/2, extracellular signal-regulated kinase 1 and 2; ITIM, immunoreceptor tyrosine-based motifs; Src, Src kinases.
2. Orexins and Digestive Cancers
2.1. Colon Cancer
Among digestive cancers, colorectal cancer (CRC) represents the third most common cancer world-wide and the third highest cause of cancer-related mortality, which is responsible of about 10% of total cancer death [9]. CRC development resulted from the transformation of normal epithelium to adenoma and then adenocarcinoma. This transformation was associated with multiple genetic and epigenetic alterations consisting of chromosomal instability and microsatellite instability that led to damage in tumor suppressor genes such as Apc, Kras, Smad, Cdc, Tp53… [10] which dysregulated important intracellular signaling pathways. Moreover, epigenetic alterations (CpG methylation, histones acetylation) also caused a gene dysregulation [10]. If surgery was often the first line of treatment for early-stage cancers, then in more advanced metastatic cancers, chemotherapy based on fluoropyrimidines such as 5-fluorouracil (5-FU), oxaliplatin and irinotecan was proposed [11]. It should be noted that recent treatments based on immunotherapy (anti-PDL-1) and targeted drug therapy can be associated with chemotherapy [12]. Among genetic/epigenetic remodeling in cancer cells, the aberrant expression or the inhibition of various proteins’ expression had a direct impact on cancer cells in terms of proliferation, apoptosis, cell signaling pathways, etc., but also opened the door to the identification of new targets that may lead to new therapeutic approaches. GPCRs represented a class of surface proteins whose expression was modulated in cancer cells by underexpression or overexpression [13]. Moreover, these GPCRs were involved in many important signaling pathways able to play a role in cancer cell proliferation, metabolism and metastasis [14]. In 2004, we demonstrated that OX1R was ectopically expressed in colon cancer and neuroblastoma in which the activation of these receptors by orexins induced an inhibition of cell growth [15]. The percentage of cells from colon tumors expressing OX1R was about 50 to 100% and was independent of tumor location and Duke’s stage. In contrast, OX1R was not expressed in normal colonic mucosa [16]. It should be keep in mind that neither OX2R or orexins were found in colon tumors and normal epithelium. Moreover, OX1R was also expressed in human hepatic metastasis from CRC, indicating that its expression was conserved throughout the epithelial-mesenchymal transition (EMT). OX1R was also expressed in various human colon cancer cell lines, such as HT-29, LoVo, Caco-2, SW620, etc. [16]. It is unknown why OX1R is expressed in colon cancer, although our personal data using methylase/acetylase inhibitors suggest that its expression is dependent on epigenetic regulation (unpublished data). The OX1R activation by OxA or OxB induced a strong inhibition of cell growth (Figure 2) [16]. In the cell growth resulting from a balance between the cell proliferation and apoptosis, our group has demonstrated that orexins did not have any impact on cell proliferation but induced a mitochondrial apoptosis [17]. The deciphering of the mechanism of action by which orexins induced apoptosis identified a new signaling pathway, involving immunoreceptor tyrosine-based motifs (ITIM) and the tyrosine-protein phosphatase non-receptor type 11 (SHP2) (Figure 1). The interaction between orexins and OX1R in colon cancer cells induced the β/γ subunits dissociation from Gq protein, leading to phosphorylation by Src kinases of two ITIM sites present in TM2 and TM7 of the receptor [17][18]. Phosphorylated receptors were able to recruit and activate SHP2, leading to the activation of p38 mitogen-stress protein kinase via RAS/MAPK signaling pathways [19]. These activation cascades induced the translocation of the proapoptotic Bax protein in mitochondria followed by the cytochrome c release involved in apoptosome formation, which led to the activation of caspases 3 and 7, which caused cell death [17][18]. The presence of functional ITIM site in OX1R sequence was not an exceptional situation in GPCR family. Indeed, in bradykinin receptor (B2) and somatostatin receptor (sst2), the presence of ITIM sites associated with SHP2 induced an inhibition of cell proliferation [20][21], whereas in cholecystokinin B receptor (CCK2), this association led to the activation of the AKT signaling pathway [22].
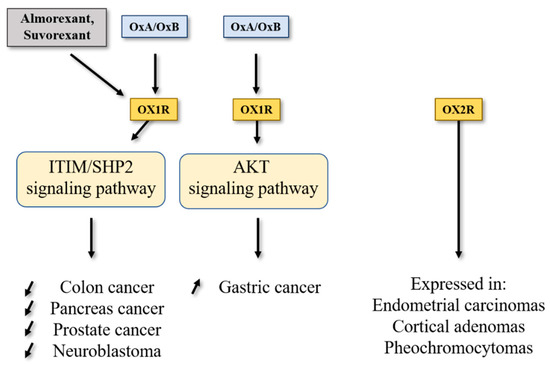
Figure 2. Impact of orexins/OXR system on cancers. SHP2, Src homology 2 (SH2) domains of SH2-containing phosphatase 2; Akt, protein kinase B; ITIM, immunoreceptor tyrosine-based motifs.
Conventional chemotherapy used in CRC treatment was mainly based on the 5-fluorouracil (5-FU) molecule either associated or not associated with other pyrimidine analogs and/or platinated agents [10]. However, the implementation of chemoresistance mechanisms (primary, before treatment, or secondary, in response to treatment) in CRC and more broadly in digestive cancers including pancreas cancer, hepatocellular cancer, gastric cancer and cholangiocarcinoma, severely limits patient remission. In the HT-29 colon cancer cell line that is resistant to 5-FU (HT-29-FU), OX1R was expressed and orexins induced cell death in these cells, demonstrating that orexin response toward apoptosis was conserved in drug-resistant cancer cells [16]. In preclinical mouse models, subcutaneous injection of colon cancer cells from LoVo or HT-29 cell lines led to the development of tumors. When OxA (or also OxB) was intraperitoneally daily injected, a strong decrease in tumor volume was observed [16]. If the OxA treatment was performed on mice with established xenografted tumors (tumor volume about 150–200 mm3), a rapid and strong reversion of tumor volume was identified demonstrating that OxA was able to reduce the established tumors [16]. Histologic analysis of control and OxA-treated tumors indicated that OX1R was similarly expressed along tumor development, showing that the OX1R expression was not modulated by OxA treatment. Moreover, large areas of cell apoptosis, revealed by activated caspase-3 staining, were observed in OxA-treated tumors [16]. In contrast, xenografts obtained with HCT-116 cells which did not express OX1R were totally insensitive to the action of OxA revealed by the absence of tumor volume inhibition [16]. Moreover, the tumor development kinetic was similar between HT-29 cells (OX1R+/+) and HCT-116 cells (OX1R−/−), indicating that: (i) OX1R expression had no impact on tumor growth in the absence of exogenous orexins; (ii) the presence (or not) of endogenous orexins had no impact on tumor growth; and (iii) the concentration of circulating orexins is too low (about 50 pM) to activate OX1R in tumors [23][24].
2.2. Pancreas Cancer
Pancreatic ductal adenocarcinoma (PDAC), which represents over 90% of pancreatic exocrine cancers, is one of the most lethal cancers, with a 5-year survival rate of about 10% [25][26]. Other rare pancreatic exocrine cancers are adenosquamous carcinoma, squamous cell carcinoma and intraductal papillary mucinous neoplasms (IPMN) including colloid carcinoma [27]. According to projections, PDAC could represent the second largest cause of cancer-related deaths in 2030 [28]. The etiology of this cancer was unknown, whereas non-specific risk factors were invoked such as smoking, age, obesity, chronic inflammation, etc. [29]. Genetic alterations characterized by PDAC were Kras mutation (over 90% of tumors were Kras mutated), P16/Cdkn2a, Tp53, Arid1a, Brca1/2, Smad4, hMlh1 and Msh2 for main mutations [30]. It should be noted that this mutation panel was modulated in other rare pancreas cancer such as IPMN, where the prevalence of Kras mutation was found, as well as the prevalence of Gnas mutation, which encoded the αs-subunit belonging to Gs protein involved in the activation of adenylyl cyclase [30]. The poor prognosis of PDAC was related to the late stage of diagnosis involving systemic metastasis for over 50% of patients. About 20% of PDAC can be surgically resectable [27], frequently associated with neoadjuvant treatment. Unfortunately, the incidence of relapse remained high (over 75%) needing chemotherapy. At this time, first line/second line chemotherapeutic treatment of advanced cancers regrouped two combinatorial regimens differing from country to country and based on Nab/GEM (Nab-paclitaxel/Gemcitabine) or FOLFIRINOX (folinic acid, 5-FU, irinotecan and oxaliplatin). However, these treatments were not well tolerated by patients and the survival gain remained relatively modest [27]. Moreover, chemoresistance resulting from metabolic reprogramming and/or genetic/epigenetic modifications of tumor and/or stromal cells appeared in PDAC [31]. In this context, identification of new targets represents an essential challenge. In 2018, it was reported that OX1R was highly expressed in 96% of 73 tested pancreatic tumors [32]. This expression was not correlated to patient age, tumor stage, tumor size, tumor differentiation and presence or not of metastasis [32]. OX1R was also expressed in precancerous lesions named intraductal papillary mucinous neoplasms (PanIN) with a gradient from low to high, dependent on PanIN grade (PanIN-1 to PanIN-3, respectively). OX1R was not expressed in normal exocrine tissue and OX2R was not expressed in pancreatic normal and tumoral tissues [32]. Moreover, OX1R was expressed in AsPC-1 cell line, which was obtained from nude mouse xenografts initiated with cells from ascites of a 62-year-old patient with PDAC [33]. Activation of OX1R by OxA in AsPC-1 cells induced a drastic inhibition of cell growth resulting in mitochondrial apoptosis, as previously described in colon cancer [16][32]. In preclinical mouse models, OxA reduced the tumor growth in nude mice subcutaneously injected with AsPC-1 cells [32]. Similarly, if isolated cells from a PDAC patient named patient-derived xenograft (PDX) were subcutaneously injected to nude mice, intraperitoneal (ip) injection of OxA also induced an inhibition of tumor growth (Figure 2). Furthermore, OxA treatment started 15–20 days after tumor development of AsPC-1 or PDX cells xenografted in nude mice; a strong and rapid decrease in established tumor volume was observed [32].
References
- Siegel, J.M.; Moore, R.; Thannickal, T.; Nienhuis, R. A brief history of hypocretin/orexin and narcolepsy. Neuropsychopharmacology 2001, 25, S14–S20.
- de Lecea, L.; Kilduff, T.S.; Peyron, C.; Gao, X.; Foye, P.E.; Danielson, P.E.; Fukuhara, C.; Battenberg, E.L.; Gautvik, V.T.; Bartlett, F.S., II; et al. The hypocretins: Hypothalamus-specific peptides with neuroexcitatory activity. Proc. Natl. Acad. Sci. USA 1998, 95, 322–327.
- Sakurai, T.; Amemiya, A.; Ishii, M.; Matsuzaki, I.; Chemelli, R.M.; Tanaka, H.; Williams, S.C.; Richardson, J.A.; Kozlowski, G.P.; Wilson, S.; et al. Orexins and orexin receptors: A family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 1998, 92, 573–585.
- Couvineau, A.; Dayot, S.; Nicole, P.; Gratio, V.; Rebours, V.; Couvelard, A.; Voisin, T. The Anti-tumoral Properties of Orexin/Hypocretin Hypothalamic Neuropeptides: An Unexpected Therapeutic Role. Front. Endocrinol. 2018, 9, 573.
- Li, S.B.; de Lecea, L. The hypocretin (orexin) system: From a neural circuitry perspective. Neuropharmacology 2020, 167, 107993–108004.
- Couvineau, A.; Voisin, T.; Nicole, P.; Gratio, V.; Abad, C.; Tan, Y.-V. Orexins as Novel Therapeutic Targets in Inflammatory and Neurodegenerative Diseases. Front. Endocrinol. 2019, 10, 709.
- Berhe, D.F.; Gebre, A.K.; Assefa, B.T. Orexins role in neurodegenerative diseases: From pathogenesis to treatment. Pharmacol. Biochem. Behav. 2020, 194, 172929.
- Kukkonen, J.P.; Turunen, P.M. Cellular Signaling Mechanisms of Hypocretin/Orexin. Orexin Syst. Basic Sci. Role Sleep Pathol. 2021, 45, 91–102.
- Rizzo, G.; Bertotti, A.; Leto, S.M.; Vetrano, S. Patient-derived tumor models: A more suitable tool for pre-clinical studies in colorectal cancer. J. Exp. Clin. Cancer Res. 2021, 40, 178.
- Cerrito, M.; Grassilli, E. Identifying Novel Actionable Targets in Colon Cancer. Biomedicines 2021, 9, 579.
- Grothey, A.; Sargent, D.; Goldberg, R.M.; Schmoll, H.-J. Survival of Patients with Advanced Colorectal Cancer Improves with the Availability of Fluorouracil-Leucovorin, Irinotecan, and Oxaliplatin in the Course of Treatment. J. Clin. Oncol. 2004, 22, 1209–1214.
- Abdul-Latif, M.; Townsend, K.; Dearman, C.; Shiu, K.-K.; Khan, K. Immunotherapy in gastrointestinal cancer: The current scenario and future perspectives. Cancer Treat. Rev. 2020, 88, 102030.
- Usman, S.; Khawer, M.; Rafique, S.; Naz, Z.; Saleem, K. The current status of anti-GPCR drugs against different cancers. J. Pharm. Anal. 2020, 10, 517–521.
- Arang, N.; Gutkind, J.S. G Protein-Coupled receptors and heterotrimeric G proteins as cancer drivers. FEBS Lett. 2020, 594, 4201–4232.
- Rouet-Benzineb, P.; Rouyer-Fessard, C.; Jarry, A.; Avondo, V.; Pouzet, C.; Yanagisawa, M.; Laboisse, C.; Laburthe, M.; Voisin, T. Orexins Acting at Native OX1 Receptor in Colon Cancer and Neuroblastoma Cells or at Recombinant OX1 Receptor Suppress Cell Growth by Inducing Apoptosis. J. Biol. Chem. 2004, 279, 45875–45886.
- Voisin, T.; El Firar, A.; Fasseu, M.; Rouyer-Fessard, C.; Descatoire, V.; Walker, F.; Paradis, V.; Bedossa, P.; Henin, D.; Lehy, T.; et al. Aberrant Expression of OX1 Receptors for Orexins in Colon Cancers and Liver Metastases: An Openable Gate to Apoptosis. Cancer Res. 2011, 71, 3341–3351.
- Voisin, T.; El Firar, A.; Rouyer-Fessard, C.; Gratio, V.; Laburthe, M. A hallmark of immunoreceptor, the tyrosine-based inhibitory motif ITIM, is present in the G protein-coupled receptor OX1R for orexins and drives apoptosis: A novel mechanism. FASEB J. 2008, 22, 1993–2002.
- El Firar, A.; Voisin, T.; Rouyer-Fessard, C.; Ostuni, M.A.; Couvineau, A.; Laburthe, M. Discovery of a functional immunoreceptor tyrosine-based switch motif in a 7-transmembrane-spanning receptor: Role in the orexin receptor OX1R-driven apoptosis. FASEB J. 2009, 23, 4069–4080.
- Laburthe, M.; Voisin, T.; El Firar, A. Orexins/hypocretins and orexin receptors in apoptosis: A mini-review. Acta Physiol. 2010, 198, 393–402.
- Duchene, J.; Schanstra, J.P.; Pecher, C.; Pizard, A.; Susini, C.; Esteve, J.-P.; Bascands, J.-L.; Girolami, J.-P. A Novel Protein-Protein Interaction between a G Protein-coupled Receptor and the Phosphatase SHP-2 Is Involved in Bradykinin-induced Inhibition of Cell Proliferation. J. Biol. Chem. 2002, 277, 40375–40383.
- Ferjoux, G.; Lopez, F.; Esteve, J.-P.; Ferrand, A.; Vivier, E.; Vely, F.; Saint-Laurent, N.; Pradayrol, L.; Buscail, L.; Susini, C. Critical Role of Src and SHP-2 in sst2 Somatostatin Receptor-mediated Activation of SHP-1 and Inhibition of Cell Proliferation. Mol. Biol. Cell 2003, 14, 3911–3928.
- Vatinel, S.; Ferrand, A.; Lopez, F.; Kowalski-Chauvel, A.; Estève, J.-P.; Fourmy, D.; Dufresne, M.; Seva, C. An ITIM-like motif within the CCK2 receptor sequence required for interaction with SHP-2 and the activation of the AKT pathway. Biochim. Biophys. Acta (BBA) Bioenerg. 2006, 1763, 1098–1107.
- Arihara, Z.; Takahashi, K.; Murakami, O.; Totsune, K.; Sone, M.; Satoh, F.; Ito, S.; Mouri, T. Immunoreactive orexin-A in human plasma. Peptides 2001, 22, 139–142.
- Sakurai, S.; Nishijima, T.; Takahashi, S.; Yamauchi, K.; Arihara, Z.; Takahashi, K. Clinical Significance of Daytime Plasma Orexin-A-Like Immunoreactivity Concentrations in Patients with Obstructive Sleep Apnea Hypopnea Syndrome. Respiration 2004, 71, 380–384.
- Jain, T.; Dudeja, V. The war against pancreatic cancer in 2020—advances on all fronts. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 99–100.
- Neuzillet, C.; Tijeras-Raballand, A.; Bourget, P.; Cros, J.; Couvelard, A.; Sauvanet, A.; Vullierme, M.-P.; Tournigand, C.; Hammel, P. State of the art and future directions of pancreatic ductal adenocarcinoma therapy. Pharmacol. Ther. 2015, 155, 80–104.
- Cherri, S.; Noventa, S.; Zaniboni, A. Pancreatic adenocarcinoma: Beyond first line, where are we? World J. Gastroenterol. 2021, 27, 1847–1863.
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921.
- Singhi, A.D.; Wood, L.D. Early detection of pancreatic cancer using DNA-based molecular approaches. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 457–468.
- Visani, M.; Acquaviva, G.; De Leo, A.; Sanza, V.; Merlo, L.; Maloberti, T.; Brandes, A.A.; Franceschi, E.; Di Battista, M.; Masetti, M.; et al. Molecular alterations in pancreatic tumors. World J. Gastroenterol. 2021, 27, 2710–2726.
- Yu, S.; Zhang, C.; Xie, K.-P. Therapeutic resistance of pancreatic cancer: Roadmap to its reversal. Biochim. Biophys. Acta (BBA) Bioenerg. 2021, 1875, 188461.
- Dayot, S.; Speisky, D.; Couvelard, A.; Bourgoin, P.; Gratio, V.; Cros, J.; Rebours, V.; Sauvanet, A.; Bedossa, P.; Paradis, V.; et al. In vitro, in vivo and ex vivo demonstration of the antitumoral role of hypocretin-1/orexin-A and almorexant in pancreatic ductal adenocarcinoma. Oncotarget 2018, 9, 6952–6967.
- Tan, M.H.; Shimano, T.; Chu, T.M. Differential Localization of Human Pancreas Cancer-Associated Antigen and Carcinoembryonic Antigen in Homologous Pancreatic Tumoral Xenograft23. J. Natl. Cancer Inst. 1981, 67, 563–569.


