 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Diego Franco | + 2969 word(s) | 2969 | 2021-07-15 04:40:14 | | | |
| 2 | Lindsay Dong | Meta information modification | 2969 | 2021-09-03 11:33:39 | | |
Video Upload Options
microRNAs represent the most studied type of small ncRNAs and it has been demonstrated that miRNAs play essential roles in multiple biological contexts, including normal development and diseases. Cardiac arrhythmias are prevalent among humans across all age ranges, affecting millions of people worldwide. While cardiac arrhythmias vary widely in their clinical presentation, they possess shared complex electrophysiologic properties at cellular level that have not been fully studied.
1. The Electrical Components of the Adult Heart
Rhythmic contraction of the heart, leading to alternative systole and diastole contraction phases is controlled by the cardiac conduction system (CCS). The CCS is formed by slow and fast conduction pathways. The slow components are two distinct low conducting and self-firing nodes, the sinoatrial and the atrioventricular node, respectively. The sinoatrial node is located at the junction between the right superior caval vein entrance and the atrial chamber myocardium and is the main pacemaker of the heart [1]. The atrioventricular node is located at the top of the interventricular septum just at the junction between atrial and ventricular myocardium. The fast conducting components of the cardiac conduction system are exclusively located in the ventricular chambers, and are composed by the bundle of His, the left and right bundle branches, and the Purkinje fiber network [1].
At cellular level, the electrical activity of the myocardial cells is governed by an exquisite balance of inward and outward ion currents that configure the cardiac action potential. The cardiac action potential can be divided in at least four different phases. The first phase is initiated with a rapid upstroke of inward sodium currents, leading to the depolarization phase. Subsequently, the repolarization phase is initiated with fine-tuned balance of outward potassium currents, leading to phases two (ITO currents) and three (IK currents) of the cardiac action potential to finally reach the fourth phase of resting membrane potential (IK1 currents) [2][3][4]. During the plateau phase, calcium contraction coupling also takes place, by the activation of the ICa,L currents followed by mobilization of intracellular calcium from the sarcoplasmic reticulum, throughout the calcium-induced calcium release mechanism [5][6][7]. This general configuration of the cardiac action potential, although applicable to all cardiomyocytes, displays subtle variations on distinct cardiac regions. Importantly, such variability is due to distinct molecular substrates governing such events, such as for example, the upstroke phase in the cardiac action potential of the cardiac conduction system is governed by cation channels, with limited contribution of the sodium channels [3][4]. In addition to the distinct functional properties of the cardiac conduction system, the working myocardium also display significant differences between each cardiac regions—e.g., between atrial and ventricular myocardium—and also within the ventricular myocardium itself—e.g., epicardial vs. endocardial configurations. Such regional differences are mainly motivated by regional differences in the relative contribution of the outward potassium channels governing the rapid (ITO; IKur), plateau (IKr; IKs), and final (IKs; IK1) phases of the cardiac action potential repolarization as well as by the L-type calcium channels in the plateau phase. Finally, it is important to highlight that there are notable species-specific differences in the contribution of the discrete ion currents to the final configuration of the cardiac action potential. Such differences are particularly applicable to the repolarization phase in distinct experimental models such as rat, mouse, pig, and zebrafish as compare to humans, as widely documented elsewhere [8][9][10][11][12][13][14].
2. Role of ncRNAs in the Cardiac Action Potential
2.1. ncRNAs in the Upstroke Phase (INa Currents)
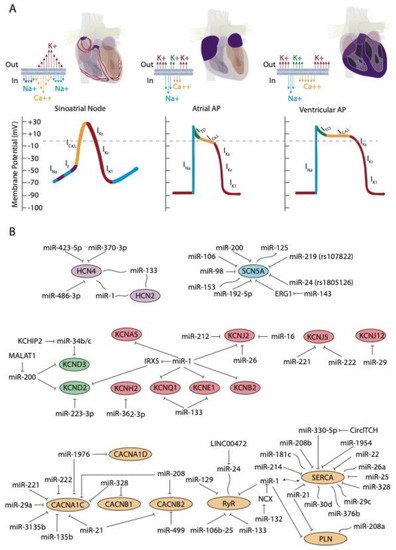
The upstroke phase of the cardiac action potential in fast conducting cells, i.e., atrial and ventricular myocytes, is primarily modulated by the fast INa current (NaV1.5) with a smaller contribution of Nav1.8. Importantly, the function of the pore-forming Nav1.5 channel is also modulated by ancillary subunits such as Navβ1-Navβ4 (Figure 1A). SCN5A encodes the voltage-gated Na+ channel NaV1.5. Mutations in SCN5A are associated to inherited arrhythmias and cardiomyopathy [15][16][17][18]. Moreover, single-nucleotide polymorphisms (SNPs) linked to SCN5A splicing, localization, and function are also associated to sudden cardiac death [19][20]. SCN10A encodes the voltage-gated Na+ channel NaV1.8. Importantly, mutations in SCN10A have also been linked to sudden unexplained death [21], atrial fibrillation [22][23], and Brugada syndrome [20][24][25]. Furthermore, SCN5A and SCN10A share common regulatory elements that are relevant for cardiac function [26].
| Current | microRNA | Gene | Function | Reference |
|---|---|---|---|---|
| INa | miR-98, miR-106, miR-200, miR-219, miR-125, miR-153 | SCN5A | INa ↑/INa ↓ | [29][30] |
| miR-192-5p | INa ↓ | [31] | ||
| miR-200c | - | [32] | ||
| miR-143 | INa ↓ | [33] | ||
| miR-24 | INa ↓ | [34] | ||
| If | miR-423-5p | HCN4 | If ↓ | [35] |
| miR-370-3p | If ↓ | [36] | ||
| miR-486-3p | If ↓ | [37] | ||
| miR-1, miR-133 | If ↑ | [38][39][40][41] | ||
| miR-1, miR-133 | HCN2 | If ↑ | [38][39][40][41] | |
| ITO | miR-1 | KCND2 | ITO ↓ | [42] |
| miR-223-3p | ITO ↓ | [43] | ||
| miR-34b/c | ITO = | [44] | ||
| miR-200 | ITO ↓ | [45] | ||
| miR-200 | KCND3 | ITO ↓ | [45] | |
| IKur | miR-1 | KCNA5 | IKur ? | [46] |
| IKr | miR-134, miR-103a-1, miR-143, miR-3619 | hERG | IKr ↓ | [47] |
| IKS | miR-1, miR-133 | KCNE1 | IKS ↓ | [48][49] |
| miR-1, miR-133 | KCNQ1 | IKS ↓ | [48] | |
| miR-1, miR-133 | KCNB2 | IKS ↓ | [49] | |
| IK1 | miR-1 | KCNJ2 | IK1 ↓/IK1 ↑ | [50][51][52] |
| miR-16 | IK1 ↓ | [53] | ||
| miR-26 | IK1 ↑ | [54][55] | ||
| miR-212 | IK1 ↓ | [56][57] | ||
| miR-29 | KCNJ12 | IK1 ↓ | [58] | |
| miR-221/222 | KCNJ5 | IK1 ↓ | [59] | |
| ICa,L | miR-328 | CACNA1C | ICa,L ↓ | [60] |
| miR-21, miR-208b | ICa,L ↓ | [61][62] | ||
| miR-20a, miR-3135b | ICa,L ↓ | [63] | ||
| miR-499 | ICa,L ↓ | [64] | ||
| miR-135b | ICa,L ↓ | [65] | ||
| miR-221/222 | ICa,L ↓ | [59] | ||
| miR-328 | CACNB1 | ICa,L ↓ | [60] | |
| miR-21, miR-208b | CACNB2 | ICa,L ↓ | [61][62] | |
| miR-499 | ICa,L ↓ | [64] | ||
| miR-329 | ICa,L ↓ | [64] | ||
| CICR | miR-106b | RYR2 | - | [66][67] |
| miR-129 | [68] | |||
| miR-1, miR-133 | [69][70] | |||
| miR-23 | [71][72] | |||
| miR-25 | SERCA2A | - | [73][74] | |
| miR-328 | [75][76] | |||
| miR-29c | [77] | |||
| miR-21 | [78] | |||
| miR-208b | [62] | |||
| miR-22 | [79] | |||
| miR-214 | [80] | |||
| miR-1954 | [81] | |||
| miR-376b, miR-1, miR-26a, miR-30d, miR-181 | [82] | |||
| miR-1, miR-21 | PLN | - | [83] | |
| miR-208a | [84] | |||
| miR-132 | NCX1 | - | [85] | |
| miR-1 | [80] |
| Gene | Disease | Alteration | Mir Related | Reference |
|---|---|---|---|---|
| SCN5A | Inherited arrhythmias and cardiomyopathy | Mutation | - | [15][16][17][18] |
| Sudden death | SNPs | - | [19][20] | |
| Brugada syndrome | SNPs/↓ expression | miR-219 | [29][30] | |
| Atrial fibrillation | ↓ expression | miR-192-5p | [31] | |
| Heart failure | SNPs/↓ expression | miR-24 | [34] | |
| SCN10A | Sudden death | Mutation | - | [21] |
| Atrial fibrillation | - | [22][23] | ||
| Brugada syndrome | - | [86][24][25] | ||
| HCN4 | Bradycardia | ↓ expression | miR-423-5p, miR-370-3p | [35][36] |
| Age atrial fibrillation | ↑ expression | miR-1, miR-133 | [38] | |
| Myocarial infarction | ↑ expression | miR-1, miR-133 | [39][40] | |
| HCN2 | Age atrial fibrillation | ↑ expression | miR-1, miR-133 | [38] |
| Myocardial infarction | ↑ expression | miR-1, miR-133 | [39][40] | |
| KCND2 | Sudden death | ↓ expression | miR-1 | [42] |
| Acute myocardial infarction | ↓ expression | miR-223-3p | [43] | |
| Myocardial infarction | ↓ expression | miR-200c | [45] | |
| KCNH2 | LQT syndrome (type 2) | Mutation | - | [87] |
| Heart failure | ↓ expression | miR362-3p | [88] | |
| KCNE2 | ||||
| LQT syndrome (type 6) | ||||
| LQT syndrome (type 1) | LQT syndrome (type 1) | ↓ expression | - | [89][90][91] |
| Atrial fibrillation | ↓ expression | miR-1 | [49] | |
| KCNB2 | ||||
| Atrial fibrillation | Myocardial infarction | ↓/↑ expression | miR-1, miR-16 | [51][54] |
| Atrial fibrillation | ↑ expression | miR-1, miR-26 | [50][52][55] | |
| Heart failure | ↓ expression | miR-212 | [57] | |
| KCNJ12 | Myocardial infarction | ↓ expression | miR-29 | [58] |
| KCNJ5 | Atrial fibrillation | ↓ expression | miR-221/222 | [59] |
| CACNA1C | Atrial fibrillation | ↓ expression | miR-221/222 | [59] |
| miR-328 | [60] | |||
| miR-21 | [61] | |||
| miR-208b | [62] | |||
| miR-29b, miR-3135b | [63] | |||
| CACNB2 | Atrial fibrillation | ↓ expression | miR-21 | [61] |
| miR-208b | [62] | |||
| miR-499, miR-329 | [64] | |||
| RYR2 | Atrial fibrillation | ↑ expression | miR-106b-25 | [66][67] |
| miR-106a, miR-93 | [67][92] | |||
| miR-129* | [68] | |||
| miR-1*, miR133* | [69][70] | |||
| miR-24* | [71][72] | |||
| SERCA2A | Atrial fibrillation | ↓ expression | miR-25 | [73][74] |
| miR-328 | [75][76] | |||
| miR-29c | [77] | |||
| miR-21*, miR-208b*, | [78][62] | |||
| miR-214*, miR-1954*, | [80][81] | |||
| miR-376b, miR-1*, | [82] | |||
| miR-26a*, miR-30d*, | [82] | |||
| miR-181c* | [82] | |||
| miR-330-5p* | [93] | |||
| PLN | Cardiac arrhythmias | - | miR-1, miR-21 | [71] |
| miR-208a* | [72] | |||
| NCX1 | Cardiac arrhythmias | - | miR-132 | [85] |
| miR-1 | [80] |
2.2. ncRNAs in the Upstroke Phase (If Current)
The hyperpolarization-activated cyclic nucleotide-gated (HCN) channels are the structural pore-forming subunits governing this current, with four HCN isoforms known (HCN1-4), among which HCN4 is the most highly expressed in the sinoatrial and atrioventricular nodes (Figure 1A).
D’Souza et al. [35] reported a direct biochemical interaction between miR-423-5p and HCN4 and they further demonstrated that miR-423-5p contributes to training-induced bradycardia by targeting HCN4. Thus miR-423-5p modulates the If current and the heart rate in mice. Yanni et al. [36] reported direct interaction between miR-370-3p and HCN4 (Figure 1B).
Indirect evidence on the role of miR-1 and miR-133 regulating HCN isoforms have been also reported. Inversed expression patterns of HCN2 and HCN4 (upregulated) and miR-1 and miR-133 (downregulated) have been reported in age-associated atrial fibrillation [38] myocardial infarction (MI) [39][40], and exercise training [41], yet it remains to be established if these microRNAs can direct target HCN isoforms (Figure 1B).
Similarly, no evidence has been reported to date on the direct functional role of microRNAs regulating HCN4 in arrhythmogenic syndromes, supporting the notion that additional studies are required in this context. A summary of the microRNA interaction with the HCN channels is provided on Table 1, while their links to distinct cardiac diseases is provided on Table 2.
2.3. ncRNAs in Sodium Channel Interacting Proteins
Calmodulin has been extensively reported to directly interact with NaV1.5 (SCN5A) sodium channel and thus to modulate its function [94][95][96][97][98][99][100][101]. Although distinct microRNAs such as miR-1 [102], let-7a [103], miR-625-5p [104], miR-525-5p [105], miR-338-5p [106], miR-185 [107], miR-145 [108], miR-30b-5p [109], and miR-675 [110] have been reported to modulate calmodulin expression, these reports exclusively describe their functional role in cardiac hypertrophy and failed to provide a direct link to sodium channel regulation. To date, the only report linking microRNAs, i.e., miR-26a, and cardiac arrhythmias, i.e., atrial fibrillation, is reported by Qi et al. [111]. Thus, the plausible contribution to sodium channel function by calmodulin interactive protein remains elusive.
3. Role of ncRNAs in Cardiac Repolarization
3.1. ncRNAs in the Early Repolarization (ITO Transient Outward K+ Current)
After cardiac depolarization, the early repolarization process is governed by cardiac transient outward potassium current (ITO). ITO is rapidly activated after a fast increase of the membrane potential, where a short-lived, hyperpolarizing outward K+ current (ITO) makes K+ ions from inside the cells to flow to the extracellular space, causing the transmembrane voltage to decrease. ITO is then quickly deactivated, stopping the repolarization and ending the first phase of the action potential [112].
Several microRNAs have been described to be involved in the regulation of these channels. In particular, in 2007, Zhao and co-workers [42], demonstrated that Kcnd2 is positively regulated by miR-1, through Irx5 inhibition in mice, thus altering the endocardial to epicardial transmural gradient controlled by Kcnd2 within the ventricular cardiomyocytes and thus resulting in ventricular repolarization abnormalities. Kcnd2 is also regulated by miR-223-3p, a microRNA that is remarkably upregulated in a rat model of acute MI and consequently, Kv4.2 protein levels and ITO density were significantly decreased [43] (Figure 1B). Such impaired modulation of Kv4.2 protein expression and thus of ITO current can cause prolongation of the action potential duration and thus promote arrhythmias.
3.2. ncRNAs in the Plateau Phase and Terminal Repolarization (IKr, IKs, IKur K+ Current)
3.3. ncRNAs Modulating the Ultra-Rapid Delayed Rectifier K+ Current (IKur)
3.4. ncRNAs Modulating the Rapid Delayed Rectifier K+ Current (IKr)
IKr currents are governed by hERG channels, also known as Kv11.1 [115]. As an homolog of the Drosophila “ether-a-go-go” (EAG) potassium channel, hERG was first cloned in the brain [116]. hERG channels are encoded by KCNH2 and mutations in KCNH2 have been associated to long QT syndrome (type 2; LQTS2) [87]. Ancillary MiRP1 (or KCNE2) subunits, that constitute single transmembrane protein homologous to KCNE1, was shown to associate with HERG channels and modulate IKr biophysical properties [117]. Mutations in KCNE2 have also been associated to long QT syndrome (type 6; LQTS6) [117].
3.5. ncRNAs Modulating the Slow Delayed Rectifier K + Current (IKs)
To date, scarce evidence is available regarding the functional impact of ncRNAs in IKs current modulation. Li et al. [48] examine miR-1/miR-133 levels, the potassium channel KCNE1 and KCNQ1 levels and IKs current in cardiac progenitor cells (CPCs) of normal human hearts. These authors observed that human CPCs expressed KCNE1 and KCNQ1 and possessed functional IKs currents (Figure 1B).
3.6. ncRNAs in the Resting Membrane Potential (IK1 Current and Na,K ATPase)
Several microRNAs have been reported to modulate IK1 current in distinct biological contexts. miR-1 levels are increased in patients with coronary artery diseases (CAD) and also in an experimental rat model of MI. In this context, miR-1 silences KCNJ2 protein expression, and also GJA1, by directly targeting their 3´UTRs, respectively [51] (Figure 1B). On the contrary hand, miR-1 levels are greatly reduced in human AF as well as in AF experimental models, contributing to upregulation of Kir2.1 subunit, leading thus to increased IK1, being this upregulation of inward-rectifier currents important for AF maintenance [50][52]. Additionally, miR-16 overexpression suppress KCNJ2/Kir2.1 expression in a rat experimental model of MI [53].
A summary of the microRNA interaction with the potassium channels is provided on Table 1, while their links to distinct cardiac diseases is provided on Table 2.
4. Role of ncRNAs in Conduction Contraction Coupling
4.1. ncRNAs in Calcium Currents (ICa,L Current)
Transgenic mice overexpressing miR-1 resulted in severe electrophysiological defects, causing atrioventricular block [118]. Molecular analysis demonstrates that several key components contributing to the electrical wiring of the heart were altered, such as Cx43 and Kir2.1. Electrophysiological studies revealed that ICa and IK1 currents were decreased. Knockdown of miR-1 overexpression using LNA-anti-miR-1 administration reversed such electrophysiological alteration, demonstrating a pivotal role for miR-1 in cardiac electrophysiology and particularly in calcium homeostasis. More recently, Zhang et al. (2019) demonstrated that transgenic mice overexpressing miR-1976 directly targeted two key calcium channels (i.e., Cav1.2 and Cav1.3, encoded by CACNA1C and CACNA1D, respectively) resulting in SAN dysfunction and thus lower heart rates, a phenotype reminiscent of sick sinus node syndrome in humans [119].
4.2. The Role of ncRNAs in Calcium-Induced Calcium Release
5. Conclusions and Perspectives
References
- Van Weerd, J.H.; Christoffels, V.M. The formation and function of the cardiac conduction system. Development 2016, 143, 197–210.
- Shih, H.T. Anatomy of the action potential in the heart. Texas Heart Inst. J. 1994, 21, 30–41.
- Nerbonne, J.M.; Kass, R.S. Molecular physiology of cardiac repolarization. Physiol. Rev. 2005, 85, 1205–1253.
- Bartos, D.C.; Grandi, E.; Ripplinger, C.M. Ion channels in the heart. Compr. Physiol. 2015, 5, 1423–1464.
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195.
- Endo, M. Calcium-induced release of calcium from the sarcoplasmic reticulum. Am. J. Physiol. Cell Physiol. 2007, 592, 275–285.
- Fabiato, A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 1983, 245, C1–C14.
- Milani-Nejad, N.; Janssen, P.M.L. Small and large animal models in cardiac contraction research: Advantages and disadvantages. Pharmacol. Ther. 2014, 141, 235–249.
- Vornanen, M.; Hassinen, M. Zebrafish heart as a model for human cardiac electrophysiology. Channels 2016, 10, 101–110.
- Cheng, J. Evidences of the gender-related differences in cardiac repolarization and the underlying mechanisms in different animal species and human. Fundam. Clin. Pharmacol. 2006, 20, 1–8.
- McKinnon, D.; Rosati, B. Transmural gradients in ion channel and auxiliary subunit expression. Prog. Biophys Mol. Biol 2016, 122, 165–186.
- Barry, D.M.; Nerbonne, N.J. Myocardial potassium channels: Electrophysiological and molecular diversity. Annu. Rev. Physiol. 1996, 58, 363–394.
- Tanaka, H.; Namekata, I.; Nouchi, H.; Shigenobu, K.; Kawanishi, T.; Takahara, A. New aspects for the treatment of cardiac diseases based on the diversity of functional controls on cardiac muscles: Diversity in the excitation-contraction mechanisms of the heart. J. Pharmacol. Sci. 2009, 109, 327–333.
- Ber, D.M. Species Differences and the Role of Sodium-Calcium Exchange in Cardiac Muscle Relaxation. Ann. N. Y. Acad. Sci. 1991, 639, 375–385.
- Amin, A.S.; Tan, H.L.; Wilde, A.A.M. Cardiac ion channels in health and disease. Heart Rhythm 2010, 7, 117–126.
- Remme, C.A. Cardiac sodium channelopathy associated with SCN5A mutations: Electrophysiological, molecular and genetic aspects. J. Physiol. 2013, 591, 4099–4116.
- Amin, A.S.; Asghari-Roodsari, A.; Tan, H.L. Cardiac sodium channelopathies. Pflugers Arch. Eur. J. Physiol. 2010, 460, 223–237.
- Pérez-Agustín, A.; Pinsach-Abuin, M.L.; Pagans, S. Role of non-coding variants in brugada syndrome. Int. J. Mol. Sci. 2020, 21, 8556.
- Splawski, I.; Shen, J.; Timothy, K.W.; Lehmann, M.H.; Priori, S.; Robinson, J.L.; Moss, A.J.; Schwartz, P.J.; Towbin, J.A.; Vincent, G.M.; et al. Spectrum of mutations in Long-QT Syndrome genes: KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 2000, 102, 1178–1185.
- Bezzina, C.R.; Barc, J.; Mizusawa, Y.; Remme, C.A.; Gourraud, J.B.; Simonet, F.; Verkerk, A.O.; Schwartz, P.J.; Crotti, L.; Dagradi, F.; et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 2013, 45, 1044–1049. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3624763/pdf/nihms412728.pdf (accessed on 25 June 2021).
- Gando, I.; Williams, N.; Fishman, G.I.; Sampson, B.A.; Tang, Y.; Coetzee, W.A. Functional characterization of SCN10A variants in several cases of sudden unexplained death. Forensic Sci. Int. 2019, 301, 289–298.
- Delaney, J.T.; Muhammad, R.; Shi, Y.; Schildcrout, J.S.; Blair, M.; Short, L.; Roden, D.M.; Darbar, D. Common SCN10A variants modulate PR interval and heart rate response during atrial fibrillation. Europace 2014, 16, 485–490.
- Savio-Galimberti, E.; Weeke, P.; Muhammad, R.; Blair, M.; Ansari, S.; Short, L.; Atack, T.C.; Kor, K.; Vanoye, C.G.; Olesen, M.S.; et al. SCN10A/Nav1.8 modulation of peak and late sodium currents in patients with early onset atrial fibrillation. Cardiovasc. Res. 2014, 104, 355–363.
- Hu, D.; Barajas-Martínez, H.; Pfeiffer, R.; Dezi, F.; Pfeiffer, J.; Buch, T.; Betzenhauser, M.J.; Belardinelli, L.; Kahlig, K.M.; Rajamani, S.; et al. Mutations in SCN10A are responsible for a large fraction of cases of brugada syndrome. J. Am. Coll. Cardiol. 2014, 64, 66–79.
- Monasky, M.M.; Micaglio, E.; Vicedomini, G.; Locati, E.T.; Ciconte, G.; Giannelli, L.; Giordano, F.; Crisa, S.; Vecchi, M.; Borrelli, V.; et al. Comparable clinical characteristics in Brugada syndrome patients harboring SCN5A or novel SCN10A variants. Europace 2019, 21, 1550–1558.
- Van Den Boogaard, M.; Smemo, S.; Burnicka-Turek, O.; Arnolds, D.E.; Van De Werken, H.J.G.; Klous, P.; McKean, D.; Muehlschlegel, J.D.; Moosmann, J.; Toka, O.; et al. A common genetic variant within SCN10A modulates cardiac SCN5A expression. J. Clin. Investig. 2014, 124, 1844–1852.
- Zhao, J.; Lee, M.C.; Momin, A.; Cendan, C.M.; Shepherd, S.T.; Baker, M.D.; Asante, C.; Bee, L.; Berthy, A.; Perkins, J.R.; et al. Small RNAs control sodium channel expression, nociceptor excitability, and pain thresholds. J. Neurosci. 2010, 30, 10860–10871.
- Yan, J.; Yu, H.; Shen, J.; Han, C.; Li, C.; Shen, X.; Li, B. Early over-expressing of microRNA-145 effectively precludes the development of neuropathic mechanical hyperalgesia via suppressing Nav1.8 in diabetic rats. Pain Physician 2020, 23, E673–E686.
- Daimi, H.; Lozano-Velasco, E.; Haj Khelil, A.; Chibani, J.B.E.; Barana, A.; Amorós, I.; Gonzáles de la Fuente, M.; Caballero, R.; Aranega, A.; Franco, D. Regulation of SCN5A by microRNAs: miR-219 modulates SCN5A transcript expression and the effects of flecainide intoxication in mice. Heart Rhythm 2015, 12, 1333–1342. Available online: http://www.ncbi.nlm.nih.gov/pubmed/25701775 (accessed on 15 April 2016).
- Daimi, H.; Khelil, A.H.; Neji, A.; Ben Hamda, K.; Maaoui, S.; Aranega, A.; Chibani, J.B.; Franco, D. Role of SCN5A coding and non-coding sequences in Brugada syndrome onset: What’s behind the scenes? Biomed. J. 2019, 42, 252–260.
- Zhao, Y.; Huang, Y.; Li, W.; Wang, Z.; Zhan, S.; Zhou, M.; Yao, Y.; Zen, Z.; Hou, Y.; Chen, Q.; et al. Post-transcriptional regulation of cardiac sodium channel gene SCN5A expression and function by miR-192-5p. Biochim. Biophys. Acta 2015, 1852, 2024–2034.
- Poon, E.N.Y.; Hao, B.; Guan, D.; Li, M.J.; Lu, J.; Yang, Y.; Wu, B.; Wu, S.C.M.; Webb, S.E.; Liang, Y.; et al. Integrated transcriptomic and regulatory network analyses identify microRNA-200c as a novel repressor of human pluripotent stem cell-derived cardiomyocyte differentiation and maturation. Cardiovasc. Res. 2018, 114, 894–906.
- Li, J.; Xu, C.; Liu, Y.; Li, Y.; Du, S.; Zhang, R.; Sun, Y.; Zhang, R.; Wang, Y.; Xue, H.; et al. Fibroblast growth factor 21 inhibited ischemic arrhythmias via targeting miR-143/EGR1 axis. Basic Res. Cardiol. 2020, 115, 9.
- Zhang, X.; Yoon, J.Y.; Morley, M.; McLendon, J.M.; Mapuskar, K.A.; Gutmann, R.; Mehdi, H.; Bloom, H.L.; Dudley, S.C.; Ellinor, P.T.; et al. A common variant alters SCN5A-miR-24 interaction and associates with heart failure mortality. J. Clin. Investig. 2018, 128, 1154–1163.
- D’Souza, A.; Pearman, C.M.; Wang, Y.; Nakao, S.; Logantha, S.J.R.J.; Cox, C.; Bennett, H.; Zhang, Y.; Johnsen, A.B.; Linscheid, N.; et al. Targeting miR-423-5p Reverses Exercise Training-Induced HCN4 Channel Remodeling and Sinus Bradycardia. Circ. Res. 2017, 121, 1058–1068.
- Yanni, J.; D’Souza, A.; Wang, Y.; Li, N.; Hansen, B.J.; Zakharkin, S.O.; Smith, M.; Hayward, C.; Whitson, B.; Mohler, P.J.; et al. Silencing miR-370-3p rescues funny current and sinus node function in heart failure. Sci. Rep. 2020, 10, 11279.
- Petkova, M.; Atkinson, A.J.; Yanni, J.; Stuart, L.; Aminu, A.J.; Ivanova, A.D.; Pustovit, K.B.; Geragthy, C.; Feather, A.; Li, N.; et al. Identification of Key Small Non-Coding MicroRNAs Controlling Pacemaker Mechanisms in the Human Sinus Node. J. Am. Heart Assoc. 2020, 9, e016590.
- Li, Y.D.; Hong, Y.F.; Yusufuaji, Y.; Tang, B.P.; Zhou, X.H.; Xu, G.J.; Li, J.X.; Sun, L.; Zhang, J.H.; Xin, Q.; et al. Altered expression of hyperpolarization-activated cyclic nucleotide-gated channels and microRNA-1 and -133 in patients with age-associated atrial fibrillation. Mol. Med. Rep. 2015, 12, 3243–3248.
- Suffredini, S.; Stillitano, F.; Comini, L.; Bouly, M.; Brogioni, S.; Ceconi, C.; Ferrati, R.; Mugelli, A.; Cerbai, E. Long-term treatment with ivabradine in post-myocardial infarcted rats counteracts f-channel overexpression. Br. J. Pharmacol. 2012, 165, 1457–1466.
- Yu, H.D.; Xia, S.; Zha, C.Q.; Deng, S.B.; Du, J.L.; She, Q. Spironolactone Regulates HCN Protein Expression Through Micro-RNA-1 in Rats with Myocardial Infarction. J. Cardiovasc. Pharmacol. 2015, 65, 587–592.
- D’souza, A.; Bucchi, A.; Johnsen, A.B.; Logantha, S.J.R.J.; Monfredi, O.; Yanni, J.; Prehar, S.; Hart, G.; Cartwright, E.; Wisloff, U.; et al. Exercise training reduces resting heart rate via downregulation of the funny channel HCN4. Nat. Commun. 2014, 5, 3775.
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of Cardiogenesis, Cardiac Conduction, and Cell Cycle in Mice Lacking miRNA-1-2. Cell 2007, 129, 303–317.
- Liu, X.; Zhang, Y.; Du, W.; Liang, H.; He, H.; Zhang, L.; Pan, Z.; Li, X.; Xu, C.; Zhou, Y.; et al. MiR-223-3p as a Novel MicroRNA Regulator of Expression of Voltage-Gated K + Channel Kv4.2 in Acute Myocardial Infarction. Cell Physiol. Biochem. 2016, 39, 102–114.
- Nassal, D.M.; Wan, X.; Liu, H.; Maleski, D.; Ramirez-Navarro, A.; Moravec, C.S.; Ficker, E.; LAurita, K.; Deschenes, I. KChIP2 is a core transcriptional regulator of cardiac excitability. Elife 2017, 6, 1–24.
- Zhu, P.; Yang, M.; Ren, H.; Shen, G.; Chen, J.; Zhang, J.; Liu, J.; Sun, C. Long noncoding RNA MALAT1 downregulates cardiac transient outward potassium current by regulating miR-200c/HMGB1 pathway. J. Cell Biochem. 2018, 119, 10239–10249.
- Mondejar-Parreño, G.; Callejo, M.; Barreira, B.; Morales-Cano, D.; Esquivel-Ruiz, S.; Moreno, L.; Cogolludo, A.; Perez-Vizcaino, F. miR-1 is increased in pulmonary hypertension and downregulates Kv1.5 channels in rat pulmonary arteries. J. Physiol. 2019, 597, 1185–1197.
- Lian, J.; Guo, J.; Huang, X.; Yang, X.; Huang, G.; Mao, H.; Sun, H.H.; Ba, Y.; Zhou, J. miRNAs Regulate hERG. J. Cardiovasc. Electrophysiol. 2016, 27, 1472–1482.
- Li, Y.; Yang, C.M.; Xi, Y.; Wu, G.; Shelat, H.; Gao, S.; Cheng, J.; Geng, Y.J. MicroRNA-1/133 targeted dysfunction of potassium channels KCNE1 and KCNQ1 in human cardiac progenitor cells with simulated hyperglycemia. IJC 2013, 167, 1076–1078.
- Jia, X.; Zheng, S.; Xie, X.; Zhang, Y.; Wang, W.; Wang, Z.; Zhang, Y.; Wang, J.; Gao, M.; Hou, Y. MicroRNA-1 accelerates the shortening of atrial effective refractory period by regulating KCNE1 and KCNB2 expression: An atrial tachypacing rabbit model. PLoS ONE 2013, 8. Available online: https://pubmed.ncbi.nlm.nih.gov/24386485/ (accessed on 11 December 2020).
- Chinchilla, A.; Daimi, H.; Lozano-Velasco, E.; Dominguez, J.N.; Caballero, R.; Delpo, E.; Tamargo, J.; Cinca, J.; Hove-Madsen, L.; Aranega, A.; et al. PITX2 insufficiency leads to atrial electrical and structural remodeling linked to arrhythmogenesis. Circ. Cardiovasc. Genet. 2011, 4, 269–279.
- Yang, B.; Lin, H.; Xiao, J.; Lu, Y.; Luo, X.; Li, B.; Zhang, Y.; Xu, C.; Bai, Y.; Wang, H.; et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat. Med. 2007, 13, 486–491.
- Girmatsion, Z.; Biliczki, P.; Bonauer, A.; Wimmer-Greinecker, G.; Scherer, M.; Moritz, A.; Bukowska, A.; Goetter, A.; Nattel, S.; Hohnloser, S.H.; et al. Changes in microRNA-1 expression and IK1 up-regulation in human atrial fibrillation. Heart Rhythm 2009, 6, 1802–1809.
- Li, X.; Hu, H.; Wang, Y.; Xue, M.; Li, X.; Cheng, W.; Xuan, Y.; Yin, J.; Yang, N.; Yan, S. Valsartan ameliorates KIR2.1 in rats with myocardial infarction via the NF-κB-miR-16 pathway. Gene 2016, 590, 201–209.
- Luo, X.; Pan, Z.; Shan, H.; Xiao, J.; Sun, X.; Wang, N.; Lin, X.; Xiao, L.; Maguy, A.; Qi, X.Y.; et al. MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. JCI 2013, 123, 1939–1951. Available online: https://pubmed.ncbi.nlm.nih.gov/23543060/ (accessed on 11 December 2020).
- Qi, X.Y.; Huang, H.; Ordog, B.; Luo, X.; Naud, P.; Sun, Y.; Wu, C.T.; Dawson, K.; Tadevosyan, A.; Chen, Y.; et al. Fibroblast inward-rectifier potassium current upregulation in profibrillatory atrial Remodeling. Circ. Res. 2015, 116, 836–845.
- Thum, T.; Galuppo, P.; Wolf, C.; Fiedler, J.; Kneitz, S.; Van Laake, L.W.; Doevendans, P.A.; Mummery, C.L.; Borlak, J.; Haverich, A.; et al. MicroRNAs in the human heart: A clue to fetal gene reprogramming in heart failure. Circulation 2007, 116, 258–267.
- Goldoni, D.; Yarham, J.M.; McGahon, M.K.; O’Connor, A.; Guduric-Fuchs, J.; Edgar, K.; McDonald, D.M.; Simpson, D.A.; Collins, A. A novel dual-fluorescence strategy for functionally validating microRNA targets in 3′ untranslated regions: Regulation of the inward rectifier potassium channel Kir2.1 by miR-212. Biochem. J. 2012, 448, 103–113.
- Zhang, X.J.; Liao, C.X.; Sun, K.J.; Liu, L.L.; Xu, D.Y. A soluble epoxide hydrolase inhibitor upregulated kcnj12 and kcnip2 by downregulating microrna-29 in a mouse model of myocardial infarction. Heart Surg. Forum. 2020, 23, E579–E585.
- Binas, S.; Knyrim, M.; Hupfeld, J.; Kloeckner, U.; Rabe, S.; Mildenberger, S.; Quarch, K.; Stratz, N.; Misiak, D.; Gekle, M.; et al. miR-221 and -222 target CACNA1C and KCNJ5 leading to altered cardiac ion channel expression and current density. CLMS 2019, 77, 903–918.
- Lu, Y.; Zhang, Y.; Wang, N.; Pan, Z.; Gao, X.; Zhang, F.; Zhang, Y.; Shan, H.; Luo, X.; Bai, Y.; et al. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation 2010, 122, 2378–2387.
- Barana, A.; Matamoros, M.; Dolz-Gaitón, P.; Pérez-Hernández, M.; Amorós, I.; Núñez, M.; Sacristan, S.; Pedraz, A.; Pinto, A.; Fernandez-Aviles, F.; et al. Chronic atrial fibrillation increases MicroRNA-21 in human atrial myocytes decreasing L-type calcium current. Circ. Arrhythmia Electrophysiol. 2014, 7, 861–868.
- Cañón, S.; Caballero, R.; Herraiz-Martínez, A.; Pérez-Hernández, M.; López, B.; Atienza, F.; Jalife, J.; Hove-Madsen, L.; Delpon, E.; Bernard, A. miR-208b upregulation interferes with calcium handling in HL-1 atrial myocytes: Implications in human chronic atrial fibrillation. J. Mol. Cell Cardiol. 2016, 99, 162–173.
- Zhao, Y.; Yuan, Y.; Qiu, C. Underexpression of CACNA1C caused by overexpression of microRNA-29a underlies the pathogenesis of atrial fibrillation. Med. Sci. Monit. 2016, 22, 2175–2181.
- Ling, T.Y.; Wang, X.L.; Chai, Q.; Lu, T.; Stulak, J.M.; Joyce, L.D.; Daly, R.C.; Greason, K.L.; Wu, L.Q.; Shen, W.K.; et al. Regulation of cardiac CACNB2 by microRNA-499: Potential role in atrial fibrillation. BBA Clin. 2017, 7, 78–84.
- Chu, Q.; Li, A.; Chen, X.; Qin, Y.; Sun, X.; Li, Y.; Yue, E.; Wang, C.; Ding, X.; Yan, Y.; et al. Overexpression of miR-135b attenuates pathological cardiac hypertrophy by targeting CACNA1C. Int. J. Cardiol. 2018, 269, 235–241.
- Liu, Z.; Tao, B.; Fan, S.; Pu, Y.; Xia, H.; Xu, L. MicroRNA-145 protects against myocardial ischemia reperfusion injury via CaMKII-mediated antiapoptotic and anti-inflammatory pathways. Oxid. Med. Cell Longev. 2019, 10, 8948657.
- Chiang, D.Y.; Kongchan, N.; Beavers, D.L.; Alsina, K.M.; Voigt, N.; Neilson, J.R.; Jakob, H.; Martin, J.F.; Dobrev, D.; Wehrens, X.H.T.; et al. Loss of MicroRNA-106b-25 cluster promotes atrial fibrillation by enhancing ryanodine receptor type-2 expression and calcium release. Circ. Arrhythmia Electrophysiol. 2014, 7, 1214–1222.
- Li, Q.; Qin, M.; Tan, Q.; Li, T.; Gu, Z.; Huang, P.; Ren, L. MicroRNA-129-1-3p protects cardiomyocytes from pirarubicin-induced apoptosis by down-regulating the GRIN2D-mediated Ca2+ signalling pathway. J. Cell Mol. Med. 2020, 24, 2260–2271.
- Belevych, A.E.; Sansom, S.E.; Terentyeva, R.; Ho, H.T.; Nishijima, Y.; Martin, M.M.; Jindal, H.K.; Rochira, J.A.; Kunimoto, Y.; Abdellatif, M.; et al. MicroRNA-1 and -133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PLoS ONE 2011, 6, e28324.
- Terentyev, D.; Belevych, A.E.; Terentyeva, R.; Martin, M.M.; Malana, G.E.; Kuhn, D.E.; Abdellatif, M.; Feldman, D.S.; Elton, T.S.; Gyorke, S. MiR-1 Overexpression Enhances Ca2+ release and Promotes Cardiac Arrhythmogenesis by Targeting PP2A Regulatory Subunit B56α and Causing CaMKII-Dependent Hyperphosphorylation of RyR2. Circ Res. 2009, 104, 514–521.
- Li, R.C.; Tao, J.; Guo, Y.B.; Wu, H.D.; Liu, R.F.; Bai, Y.; LV, Z.Z.; Luo, G.Z.; Li, L.L.; Wang, M.; et al. In vivo suppression of microRNA-24 prevents the transition toward decompensated hypertrophy in aortic-constricted mice. Circ. Res. 2013, 112, 601–605.
- Wang, L.-Y.; Shen, H.; Yang, Q.; Min, J.; Wang, Q.; Xi, W.; Yin, L.; Le, S.G.; Zhang, Y.F.; Xiao, J.; et al. LncRNA-LINC00472 contributes to the pathogenesis of atrial fibrillation (Af) by reducing expression of JP2 and RyR2 via miR-24. Biomed. Pharmacother. 2019, 120, 109364.
- Jeong, D.; Yoo, J.; Lee, P.; Kepreotis, S.V.; Lee, A.; Wahlquist, C.; Brown, B.D.; Kho, C.; Mercola, M.; Hajjar, R.J. miR-25 Tough Decoy Enhances Cardiac Function in Heart Failure. Mol. Ther. 2018, 7, 718–729.
- Wahlquist, C.; Jeong, D.; Rojas-Muñoz, A.; Kho, C.; Lee, A.; Mitsuyama, S.; van Mil, A.; Park, W.J.; Sluijter, J.P.G.; Doevendans, P.A.F.; et al. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature 2014, 508, 531–535.
- Li, C.; Li, X.; Gao, X.; Zhang, R.; Zhang, Y.; Liang, H.; Xu, C.; Du, W.; Zhang, Y.; Liu, X.; et al. MicroRNA-328 as a regulator of cardiac hypertrophy. Int. J. Cardiol. 2014, 173, 268–276.
- Zheng, X.; Hu, X.; Ge, T.; Li, M.; Shi, M.; Luo, J.; Lai, H.; Nie, T.; Li, F.; Li, H. MicroRNA-328 is involved in the effect of selenium on hydrogen peroxide-induced injury in H9c2 cells. J. Biochem. Mol. Toxicol. 2017, 31, 3–9.
- Williams, A.L.; Walton, C.B.; MacCannell, K.A.; Avelar, A.; Shohet, R.V. HIF1 regulation of miR-29c impairs SERCA2 expression and cardiac contractility. AJPheart. 2019, 53, 21–25. Available online: http://doi.org/10.1152/ajpheart.00617.2018 (accessed on 25 June 2021).
- Mayourian, J.; Ceholski, D.K.; Gorski, P.A.; Mathiyalagan, P.; Murphy, J.F.; Salazar, S.I.; Stillitano, F.; Hare, J.M.; Sahoo, S.; Hajjar, R.J.; et al. Exosomal microRNA-21-5p Mediates Mesenchymal Stem Cell Paracrine Effects on Human Cardiac Tissue Contractility. Circ. Res. 2018, 122, 933–944.
- Gurha, P.; Abreu-Goodger, C.; Wang, T.; Ramirez, M.O.; Drumond, A.L.; Van Dongen, S.; Chen, Y.; Bartonicek, N.; Enright, A.J.; Lee, B.; et al. Targeted deletion of MicroRNA-22 promotes stress-induced cardiac dilation and contractile dysfunction. Circulation 2012, 125, 2751–2761.
- Melo, S.F.S.; Barauna, V.G.; Neves, V.J.; Fernandes, T.; Lara, L.d.S.; Mazzotti, D.R. Exercise training restores the cardiac microRNA-1 and -214 levels regulating Ca2+ handling after myocardial infarction. BMC Cardiovasc. Disord. 2015, 15, 4–11.
- Chiasson, V.; Takano, A.P.C.; Guleria, R.S.; Gupta, S. Deficiency of MicroRNA miR-1954 Promotes Cardiac Remodeling and Fibrosis. J. Am. Heart Assoc. 2019, 8, 1–13.
- Mishra, P.K.; Metreveli, N.; Tyagi, S.C. MMP-9 gene ablation and TIMP-4 mitigate PAR-1-mediated cardiomyocyte dysfunction: A plausible role of dicer and miRNA. Cell Biochem. Biophys. 2010, 57, 67–76.
- Soller, K.J.; Yang, J.; Veglia, G.; Bowser, M.T. Reversal of phospholamban inhibition of the sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) using short, protein-interacting RNAs and oligonucleotide analogs. J. Biol. Chem. 2016, 291, 21510–21518.
- Bedada, F.B.; Martindale, J.J.; Arden, E.; Metzger, J.M. Molecular inotropy mediated by cardiac miR-based PDE4D/PRKAR1α/phosphoprotein signaling. Sci. Rep. 2016, 6, 36803.
- Hong, S.; Lee, J.; Seo, H.H.; Lee, C.Y.; Yoo, K.J.; Kim, S.M.; Lee, S.; Hwang, K.C.; Choi, E. Na+-Ca2+ exchanger targeting miR-132 prevents apoptosis of cardiomyocytes under hypoxic condition by suppressing Ca2+ overload. Biochem. Biophys. Res. Commun. 2015, 460, 931–937.
- Lu, M.; Zhang, Q.; Deng, M.; Miao, J.; Guo, Y.; Gao, W.; Cui, Q. An analysis of human microRNA and disease associations. PLoS ONE 2008, 3, 1–5.
- Curran, M.E.; Splawski, I.; Timothy, K.W.; Vincen, G.M.; Green, E.D.; Keating, M.T. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995, 80, 795–803.
- Mourad, N.A.-R. Microrna Regulation of Herg-Related Current: Potential Role in Heart Failure-Associated Arrhythmias [Internet]. Purdue University, 2015. Available online: https://docs.lib.purdue.edu/dissertations/AAI10188989/ (accessed on 25 June 2021).
- Takumi, T.; Ohkubo, H.; Nakanishi, S. Cloning of a membrane protein that induces a slow voltage-gated potassium current. Science 1988, 242, 1042–1045.
- Barhanin, J.; Lesage, F.; Guillemare, E.; Fink, M.; Lazdunski, M.; Romey, G. K(V)LQT1 and IsK (minK) proteins associate to form the I(Ks) Cardiac Potassium Current. Nature 1996, 384, 78–80.
- Sanguinetti, M.C.; Curran, M.E.; Zou, A.; Shen, J.; Spector, P.S.; Atkinson, D.L.; Keating, M. Coaseembly of KvLQT1 and minK (IsK) proteins to form cardiac Iks potassium channel. Lett. Nat. 1996, 384, 80–83.
- Zhu, H.; Xue, H.; Jin, Q.H.; Guo, J.; Chen, Y.D. Increased expression of ryanodine receptor type-2 during atrial fibrillation by miR-106-25 cluster independent mechanism. Exp. Cell Res. 2019, 375, 113–117.
- Han, D.; Wang, Y.; Wang, Y.; Dai, X.; Zhou, T.; Chen, J.; Tao, B.; Zhang, J.; Cao, F. The Tumor-Suppressive Human Circular RNA CircITCH Sponges miR-330-5p to Ameliorate Doxorubicin-Induced Cardiotoxicity through Upregulating SIRT6, Survivin, and SERCA2a. Circ. Res. 2020, 127, E108–E125.
- Greer-Short, A.; Musa, H.; Alsina, K.M.; Ni, L.; Word, T.A.; Reynolds, J.O.; Gratz, D.; Lane, C.; El-Refaey, M.; Unudurthu, S.; et al. Calmodulin kinase II regulates atrial myocyte late sodium current, calcium handling, and atrial arrhythmia. Heart Rhythm 2020, 17, 503–511.
- Wang, Z.; Vermij, S.H.; Sottas, V.; Shestak, A.; Ross-Kaschitza, D.; Zaklyazminskaya, E.V.; Hudmon, A.; Pitt, G.S.; Rougier, J.S.; Abriel, H. Calmodulin binds to the N-terminal domain of the cardiac sodium channel Nav1.5. Channels 2020, 14, 268–286.
- Gabelli, S.B.; Boto, A.; Kuhns, V.H.; Bianchet, M.A.; Farinelli, F.; Aripirala, S.; Yoder, J.; Jakoncic, J.; Tomaselli, G.F.; Amzel, L.M. Regulation of the NaV 1.5 cytoplasmic domain by calmodulin. Nat. Commun. 2014, 5, 1–11.
- Gabelli, S.B.; Yoder, J.B.; Tomaselli, G.F.; Amzel, L.M. Calmodulin and Ca2+ control of voltage gated Na+ channels. Channels 2016, 10, 45–54.
- Johnson, C.N.; Pattanayek, R.; Potet, F.; Rebbeck, R.T.; Blackwell, D.J.; Nikolaienko, R.; Sequeria, V.; Le Meur, R.; Radwanski, P.B.; Davis, J.P.; et al. The CaMKII inhibitor KN93-calmodulin interaction and implications for calmodulin tuning of NaV1.5 and RyR2 function. Cell Calcium 2019, 82, 102063.
- Gardill, B.R.; Rivera-Acevedo, R.E.; Tung, C.C.; Van Petegem, F. Crystal structures of Ca2+—Calmodulin bound to NaV C-terminal regions suggest role for EF-hand domain in binding and inactivation. Proc Natl Acad Sci. USA 2019, 166, 10763–10772.
- Koval, O.M.; Snyder, J.S.; Wolf, R.M.; Pavlovicz, R.E.; Glynn, P.; Curran, J.; Leymaster, N.D.; Dun, W.; Wright, P.J.; Cardona, N.; et al. Ca2+/calmodulin-dependent protein kinase ii-based regulation of voltage-gated na+ channel in cardiac disease. Circulation 2012, 126, 2084–2094.
- Ashpole, N.M.; Herren, A.W.; Ginsburg, K.S.; Brogan, J.D.; Johnson, D.E.; Cummins, T.R.; Bers, D.M.; Hudmon, A. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J. Biol. Chem. 2012, 287, 19856–19869.
- Ikeda, S.; He, A.; Kong, S.W.; Lu, J.; Bejar, R.; Bodyak, N.; Lee, K.H.; Ma, Q.; Kang, P.M.; Golub, T.R.; et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol. Cell Biol. 2009, 29, 2193–2204.
- Zhou, X.; Sun, F.; Luo, S.; Zhao, W.; Yang, T.; Zhang, G.; Gao, M.; Lu, R.; Shu, Y.; Mu, W.; et al. Let-7a is an antihypertrophic regulator in the heart via targeting calmodulin. Int. J. Biol. Sci. 2017, 13, 22–31.
- Cai, K.; Chen, H. MiR-625-5p Inhibits Cardiac Hypertrophy through Targeting STAT3 and CaMKII. Hum. Gene Ther. Clin. Dev. 2019, 30, 182–191.
- Zhang, Y.; Hou, Y.; Gao, F.M.; Xiao, J.; Li, C.W.; Tang, Y.C. lncRNA GAS5 regulates myocardial infarction by targeting the miR-525-5p/CALM2 axis. J. Cell Biochem. 2019, 120, 18678–18688.
- Li, K.; Lin, Y.; Li, C. MiR-338-5p ameliorates pathological cardiac hypertrophy by targeting CAMKIIδ. Arch Pharm. Res. 2019, 42, 1071–1080.
- Kim, J.O.; Song, D.W.; Kwon, E.J.; Hong, S.E.; Song, H.K.; Min, C.K.; Kim, D.H. MiR-185 plays an anti-hypertrophic role in the heart via multiple targets in the calcium-signaling pathways. PLoS ONE 2015, 13, e0122509.
- Cha, M.J.; Jang, J.K.; Ham, O.; Song, B.W.; Lee, S.Y.; Lee, C.Y.; Park, J.H.; Lee, J.; Seo, H.H.; Choi, E.; et al. MicroRNA-145 suppresses ROS-induced Ca2+ overload of cardiomyocytes by targeting CaMKIIδ. Biochem. Biophys. Res. Commun. 2013, 435, 720–726.
- He, J.; Jiang, S.; Li, F.L.; Zhao, X.J.; Chu, E.F.; Sun, M.N.; Chen, M.Z.; Li, H. MicroRNA-30b-5p is involved in the regulation of cardiac hypertrophy by targeting CaMKIIδ. J. Investig. Med. 2013, 61, 604–612.
- Liu, P.Y.; Tian, Y.; Xu, S.Y. Mediated protective effect of electroacupuncture pretreatment by miR-214 on myocardial ischemia/reperfusion injury. J. Geriatr. Cardiol. 2014, 11, 303–310.
- Qi, X.Y.; Hassani, F.V.; Hoffmann, D.; Xiao, J.; Xiong, F.; Villeneuve, L.R.; Ljubojevic-Holzer, S.; Kamler, M.; Abu-Taha, I.; Heijman, J.; et al. Inositol Trisphosphate Receptors and Nuclear Calcium in Atrial Fibrillation. Circ. Res. 2021, 128, 619–635.
- Wettwer, E.; Amos, G.; Gath, J.; Zerkowski, H.R.; Reidemeister, J.C.; Ravens, U. Transient outward current in human and rat ventricular myocytes. Cardiovasc. Res. 1993, 27, 1662–1669.
- Feng, J.; Xu, D.; Wang, Z.; Nattel, S. Ultrarapid delayed rectifier current inactivation in human atrial myocytes: Properties and consequences. Am. J. Physiol. Heart Circ. Physiol. 1998, 275, 1–3.
- Rampe, D.; Wang, Z.; Fermini, B.; Wible, B.; Dage, R.C.; Nattel, S. Voltage- and time-dependent block by perhexiline of K+ currents in human atrium and in cells expressing a Kv1.5-type cloned channel. J. Pharmacol. Exp. Ther. 1995, 274, 444–449.
- Sanguinetti, M.C.; Jiang, C.; Curran, M.E.; Keating, M.T. A mechanistic link between an inherited and an acquird cardiac arrthytmia: HERG encodes the IKr potassium channel. Cell 1995, 81, 299–307.
- Warmke, J.W.; Ganetzky, B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci. USA 1994, 91, 3438–3442.
- Abbott, G.W.; Sesti, F.; Splawski, I.; Buck, M.E.; Lehmann, M.H.; Timothy, K.W.; Keating, M.T.; Goldstein, S.A. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 1999, 97, 175–187.
- Zhang, Y.; Sun, L.; Zhang, Y.; Liang, H.; Li, X.; Cai, R.; Wang, L.; Du, W.; Zhang, R.; Li, J.; et al. Overexpression of microRNA-1 causes atrioventricular block in rodents. Int. J. Biol. Sci. 2013, 9, 445–462.
- Zhang, J.; Wei, F.; Ding, L.; Wang, L.; Zhang, X.; Yu, L.; Liu, R.; Kuang, X.; Jiao, B.; Yang, B.; et al. MicroRNA-1976 regulates degeneration of the sinoatrial node by targeting Cav1.2 and Cav1.3 ion channels. J. Mol. Cell Cardiol. 2019, 134, 74–85.


