2. Telomeres, Telomerase, and TERT
Telomeres are ribonucleoprotein caps composed of short repetitive non-coding DNA sequences and associated proteins that protect the ends of eukaryotic chromosomes
[8]. Telomere length varies among species and serves to mitigate two biological complications. First, together with the associated Shelterin complex, telomeres help protect DNA near chromosome ends from the DNA damage response (DDR), which is induced by double-stranded breaks in the DNA and can lead to cell cycle arrest, apoptosis, or repair. Second, telomeres provide a cushion of non-coding DNA to postpone the end replication crisis, in which genome instability results from gradual shortening of the lagging strand in semi-conservative DNA replication. Due to the presence of telomeres, chromosomes can withstand 50–90 replication cycles while losing around 50–150 nucleotides per mitosis, even in the absence of telomere maintenance mechanisms
[9].
Telomerase, a multi-subunit reverse transcriptase enzyme, plays a significant role in telomere maintenance. Telomerase levels are undetectable in most somatic cells; however, this enzyme is active in certain proliferating cells, germ cells, and the majority of cancers (~90%)
[10]. The telomerase holoenzyme is composed of a catalytic protein subunit encoded by
TERT, an RNA template component (
TERC), and auxiliary proteins. TERT consists of four main domains: the TERT N-terminal domain (TEN), the TERT RNA binding domain (TRBD), a reverse transcriptase domain (RT), and a C-terminal extension domain. The TEN domain binds to the ssDNA of telomere overhangs at the 3′ end, the TRBD domain interacts with
TERC, and the RT and C-terminal extension domains are involved in catalyzing the addition of telomere repeats to the 3′ end and binding to the resultant DNA/RNA hybrid
[8][10]. In somatic cells,
TERT is transcriptionally repressed via epigenetic mechanisms, mainly in the
TERTp region
[9].
TERT is also involved in many roles independent of its telomere lengthening activity
[11]. These non-canonical functions of TERT include regulating gene expression, signal transduction, mitochondrial metabolism, and resistance to ionizing radiation
[12]. Some of the most well-studied pathways regulated by TERT include the WNT/β-catenin pathway and the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway, along with MYC, vascular endothelial growth factor (VEGF), and others
[11]. TERT is found not only in the nucleus but also in the cytoplasm and mitochondria and is involved in tumorigenesis and cancer therapy resistance independent of telomere lengthening
[12]. For example, the overexpression of mitochondrial
TERT by HCC cells increases chemotherapeutic resistance (in vitro and in vivo) by increasing the mitochondrial membrane potential and inhibiting the mitochondrial apoptotic pathway
[13]. The diverse roles of TERT are highlighted in
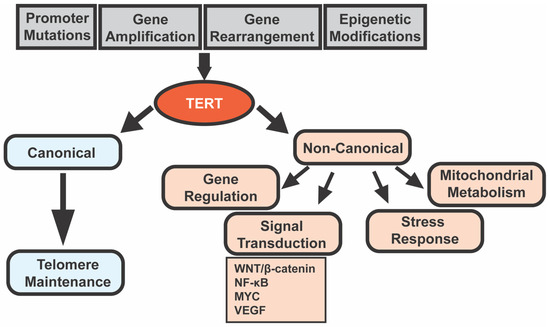
Figure 1.
Figure 1. Telomerase reverse transcriptase (
TERT) regulation and functions in cancer cells. Cancer cells may upregulate
TERT by somatic
TERTp mutations,
TERT amplification and rearrangement, and/or epigenetic modifications in
TERT and
TERTp [8]. TERT performs both canonical (telomere lengthening) and non-canonical (gene regulation, signal transduction, stress response, and mitochondrial metabolism) functions
[11][12]. Abbreviations: NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; and VEGF, vascular endothelial growth factor.
3. Telomerase and Cancer
Without a telomere maintenance mechanism, cells undergoing oncogenesis and rapid divisions gradually suffer from telomere attrition, which eventually triggers replicative senescence or apoptosis
[14]. To overcome this replication barrier caused by critical telomere length, cancer cells usually increase the TERT levels and/or TERT activity via (1)
TERTp mutations, which increase
TERT mRNA levels by creating new ETS binding motifs
[15]; (2) amplification of
TERT and/or
TERC; (3)
TERT rearrangement; and/or (4) epigenetic regulation of
TERT and its promoter
[8]. As cells in high-turnover tissues have higher telomerase activity, they may be able to stably upregulate
TERT without acquiring activating mutations like
TERTp mutations
[14]. Supporting this possibility, cancers originating from tissues with high turnover rates tend to have a lower frequency of
TERTp mutations than those from low-turnover tissues
[14].
Thus, telomerase promotes telomere maintenance, and telomere maintenance is a cancer hallmark that allows malignant cells to overcome replicative senescence. TERT also promotes cellular transformation and proliferation independent of its telomerase activity
[11]. Therefore, one would predict that loss-of-function mutations in
TERT or
TERC would inhibit tumorigenesis, while overexpression of
TERT would promote tumor formation.
Table 1 summarizes recent findings from cancer models with manipulation of
Tert or
Terc in mice or zebrafish. Supporting the aforementioned prediction,
Tert overexpression in mice leads to an increased incidence of T-cell lymphomas
[16], skin papillomas
[17], and mammary tumors
[18], depending on the promoter used to drive its expression (
Table 1).
Tert loss of function mutants display a delayed onset of mammary tumors
[19], lymphomas
[20], and early HCCs
[21] (
Table 1).
On the other hand,
Terc knockdown in mice results in the converse effect: enhancement of tumor formation (
Table 1).
Terc−/− mice treated with carbon tetrachloride (CCl
4) or diethylnitrosamine (DEN) display increased HCC “initiation foci”—microscopic lesions with atypical cytologic or architectural features that suggest malignancy but are not sufficiently well-developed to diagnose as HCC—although HCC number and size were decreased
[22]. Similarly, loss-of-function mutation of zebrafish
tert or
terc resulted in an earlier onset of spontaneous tumors, including intestinal adenocarcinomas, hematopoietic neoplasms, hepatocellular adenomas, and germ cell tumors
[23].
Why does telomerase seem to promote tumorigenesis in some animal models yet inhibit it in others? One possible explanation is that long-term inhibition of
Tert or
Terc, as in these genetic animal models, results in compensation by other telomere maintenance programs, such as alternative lengthening of telomeres (ALT). ALT is a telomerase-independent mechanism for telomere maintenance. ALT-mediated telomere synthesis occurs via homologous recombination between non-allelic telomeres, sister chromatids at telomeric sites, and/or linear or circular extrachromosomal telomeric repeats
[24]. This compensatory mechanism might not be relevant to human hepatocarcinogenesis, during which
TERT could potentially be inhibited more acutely using drugs. Inducible and/or conditional animal models in which
Tert and/or
Terc can be turned on at different time points during tumorigenesis would be helpful to dissect the importance of acute versus chronic telomerase inhibition.
Another possible reason why telomerase might inhibit tumorigenesis in some animal models while promoting it in others is related to the balance between canonical and non-canonical effects of
Tert. Laboratory mouse models have long telomeres (25–40 kb vs. 10–15 kb in humans) that are likely sufficient to permit malignant transformation even in the absence of telomerase enzyme
[12]. Therefore,
Tert overexpression and mutant phenotypes in mouse tumor models may be at least partially independent of
Tert telomere lengthening activity
[16][17][18]. As discussed in the section above, non-canonical functions of TERT tend to be tumorigenic and anti-apoptotic. Thus,
Tert deficiency in mice may delay tumorigenesis due to the resultant reduction in its non-canonical activity
[19][21].
Zebrafish have telomere lengths similar to humans (~5–15 kb)
[25] and, like humans, exhibit drastic shortening of telomeres with age
[23]; therefore, the manipulation of
tert levels is more likely to impact telomere length than in mice. Thus, the canonical effects of
tert may be relatively more important to its role in zebrafish carcinogenesis. Zebrafish
tert deficiency promotes the initiation of tumorigenesis through shortened telomeres that induce DNA damage responses, inflammation, and genetic instability
[23]. A similar phenomenon occurs in aged late-generation
Terc−/− mice, wherein shortened telomeres are associated with ulcerated skin lesions, defective wound healing, and genomic instability
[26].
These findings highlight that TERT influences tumorigenesis in both canonical and non-canonical ways. Whether the net effects of TERT are pro- or anti-tumor depends on the stage of disease, tissue type, telomere biology in different animal models, when during tumor formation TERT is manipulated, and other factors. Additional studies of vertebrate HCC models, including rigorous examination of telomere length during carcinogenesis, could help define the importance of TERT’s telomerase activity to HCC pathogenesis.
Table 1. TERT cancer models in mice and zebrafish.
| Gene (Animal) |
Site Specificity |
Expression * |
Result ** |
Ref. |
| Tert (mouse) |
Thymocytes and peripheral T cells |
+ |
↑ T-Cell Lymphomas |
[16] |
| Tert (mouse) |
Basal keratinocytes |
+ |
↑ skin papillomas (DMBA + TPA induction) |
[17] |
| Tert (mouse) |
Whole body |
+ |
↑ mammary tumors in aged females |
[18] |
| tert and terc (zebrafish) |
Neural progenitor cells |
+ |
↓ aggressiveness of RAS-mediated brain tumors |
[27] |
| Tert (mouse) |
Whole body |
− |
Delayed onset of lymphomas in EμMYC mice |
[20] |
| Tert (mouse) |
Whole body |
− |
Delayed onset of mammary tumors in PyMT mice |
[19] |
| Tert (mouse) |
Whole body |
− |
↓ HCC “initiation foci” (CCl4 induction) |
[21] |
| Terc (mouse) |
Whole body |
− |
↑ HCC “initiation foci” but ↓ HCC progression (uPA, CCl4 or DEN induction) |
[22] |
| Terc (mouse) |
Whole body |
− |
↑ tumors (lymphomas, teratocarcinomas, HCC, squamous cell carcinoma) |
[26] |
| Terc (mouse) |
Whole body |
− |
↓ skin papillomas (DMBA + TPA induction) |
[28] |
| Terc (mouse) |
Whole body |
− |
↑ epithelial cancers in TP53−/− mice |
[29] |
| Terc (mouse) |
Whole body |
− |
↑ adenoma initiation but ↓ progression in ApcMin mice |
[30] |
| terthu3430 (zebrafish) |
Whole body |
− |
Earlier onset of tumors (germ cell tumors, hematopoietic neoplasms, HCA, etc.) |
[23] |
| terthu3430 (zebrafish) |
Whole body |
− |
↑ tumor incidence and aggressiveness of melanoma model *** |
[31] |
*: +, gene overexpression; −, gene knockout or knockdown. **: ↑, increased; ↓, decreased. *** mitfa: HRAS (gives rise to melanomas) blastula cells transplanted into tert−/− casper embryos. Abbreviations: DMBA, 7,12-dimethylbenz[a]anthracene; TPA, 12-o-tetradecanoylphorbol 13-acetate; PyMT, polyomavirus middle T oncogene; CCl4, carbon tetrachloride; uPA, urokinase plasminogen activator; DEN, diethylnitrosamine; HCA, hepatocellular adenoma; Apc, adenomatous polyposis coli; ApcMin, multiple intestinal neoplasia (mutant) allele of Apc gene; and terthu3430, tert mutant line (allele hu3430) with a non-sense mutation resulting in a premature stop codon in exon 2 of tert gene.
 +1 point
+1 point