Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Enrico M. Clini | + 2474 word(s) | 2474 | 2021-08-23 11:02:23 | | | |
| 2 | Rita Xu | Meta information modification | 2474 | 2021-08-31 11:47:19 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Clini, E.M. Fibrotic Idiopathic Interstitial Lung Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/13738 (accessed on 07 February 2026).
Clini EM. Fibrotic Idiopathic Interstitial Lung Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/13738. Accessed February 07, 2026.
Clini, Enrico M.. "Fibrotic Idiopathic Interstitial Lung Disease" Encyclopedia, https://encyclopedia.pub/entry/13738 (accessed February 07, 2026).
Clini, E.M. (2021, August 31). Fibrotic Idiopathic Interstitial Lung Disease. In Encyclopedia. https://encyclopedia.pub/entry/13738
Clini, Enrico M.. "Fibrotic Idiopathic Interstitial Lung Disease." Encyclopedia. Web. 31 August, 2021.
Copy Citation
Interstitial lung diseases (ILDs) that are known as diffuse parenchymal lung diseases (DPLDs) lead to the damage of alveolar epithelium and lung parenchyma, culminating in complex inflammatory and proliferative mechanisms leading to fibrosis. ILDs that account for more than 200 different pathologies basically grouped into those that have a known cause and those where the cause is unknown, namely the idiopathic interstitial pneumonia (IIP).
lung disease
idiopathic pulmonary fibrosis
myofibroblast
extracellular matrix proteins
1. Introduction
Interstitial lung diseases (ILDs) are a heterogeneous group of pathologies that affect the lung parenchyma with wide inflammation and diffuse fibrosis [1]. Fibrotic ILDs cause the onset of progressive symptoms in patients that culminate in the decline of lung functions and often respiratory failure, which translates to a poor quality of life for patients. Interstitial lung diseases are a broad spectrum of lung pathologies that can be divided into two groups: ILDs with a known cause and ILDs without a known cause that give rise to the idiopathic form of pulmonary fibrosis [2][3]. Furthermore, the possible causes of the ILDs identified are represented by systemic diseases such as connective tissue disease [4][5] or environmental exposure to pneumotoxic drugs [6], radiation therapy [6], occupational exposures (e.g., asbestosis) [7] or allergens in the case of hypersensitivity pneumonitis [8]. The discrimination among different ILDs that present hetherogeneous inflammatory and fibrotic patterns even among patients with the same disease is critical for a more predictable prognosis and a more efficient management of the patients. Among ILDs, idiopathic pulmonary fibrosis (IPF) is the most common and severe, and for this reason the distinction between IPF and non-IPF ILDs is important given the poor prognosis in IPF compared to other fibrosing ILDs [9]. Idiopathic pulmonary fibrosis represents the most common form of idiopathic interstitial pneumonia (IIP) and is characterized by the progressive remodeling of the lung parenchyma structure, which exaggerates extracellular matrix deposition and irreversible scarring. IPF occurs primarily in older adults, with a median survival of 3 years after diagnosis, and it is diagnosed by clinicopathological criteria, including the radiographic and/or histological hallmark pattern of usual interstitial pneumonia (UIP) [10]. Prognosis remains extremely poor, since most patients die for progressive respiratory failure, often precipitated by acute events, namely, disease exacerbations [11]. Indeed, since the natural history and the course of the disease is variable among patients, the prognosis could be quite unpredictable [12]. Despite the recent introduction of the two antifibrotic drugs, namely, pirfenidone and nintedanib, that can slow down the respiratory functional decline of IPF patients according to both the real-word data and randomized controlled trials, such as CAPACITY and ASCEND, where they improved survival in patients, IPF still has a high mortality rate and survival times are quite heterogenous [3][13][14][15]. Besides the IPF, the IIPs include the non-specific interstitial pneumonia (NSIP), which is an interstitial lung disease that may be both idiopathic and secondary to connective tissue disease, toxins or other causes [16], cryptogenic organizing pneumonia (COP), also known as bronchiolitis obliterans organizing pneumonia (BOOP), that has been hypothesized to be secondary to alveolar epithelial injury due to an unknown insult, acute interstitial pneumonia (AIP) that is an extremely severe idiopathic acute interstitial disease [17], desquamative interstitial pneumonia (DIP), which is strongly associated with smoking, respiratory bronchiolitis-associated interstitial lung disease (RB-ILD), characterized by the combination of interstitial disease and respiratory bronchiolitis, and lymphocytic interstitial pneumonia (LIP) that in the majority of patients is associated with systemic autoimmune or immunodeficiency disorders, including connective tissue diseases, or in rare cases can be idiopathic [18]. The most updated version of the guidelines concerning the IIPs from the American Thoracic Society/European Respiratory Society (2013) [19] introduced some changes regarding the classification of these diseases compared to the original classification (2002) [20]. In particular, the IIPs are divided into four main groups (chronic fibrosing, smoking-related, acute/subacute, and rare) with the addition of a new disease: idiopathic pleuro parenchymal fibroelastosis [1]. Most of the ILDs have unknown etiology while there are a group of ILDs related/due to occupational and environmental exposure [21][22][23]. Although the majority of the ILDs can have unknown etiology, different works suggest that environmental factors could represent a risk factor in ILDs such as IPF since they may increase the probability of developing the disease in genetically susceptible individuals. In particular, according to the current integral model, the fibrotic pathway in IPF patients is activated by recurrent alveolar epithelium injury (e.g., environmental risks) that, together with compromised repair mechanisms of alveolar epithelium, leads to the initiation, development and progression of the disease [24][25][26]. Thus, the alveolar epithelial cells (AECs) are unable to properly respond to repetitive injuries, resulting in the loss of epithelial integrity that, together with the secretion of pro-fibrotic factors, represents the initial crucial mechanism of IPF development promoting fibroblast migration, proliferation, activation and differentiation into myofibroblasts with deposition of Extracellular Matrix (ECM) and the following distortion of the lung architecture. From a macroscopic point of view, these processes cause an increase in the stiffness of the pulmonary parenchyma, leading to the impairment of gas exchange in the alveolar district. Since the epithelial cells represent the initiator of the pulmonary fibrosis and the myofibroblasts are the key effector cells that orchestrate the progression of the disease, several recent studies aiming to clarify the molecular mechanism behind the IPF progression are focused on the identification of the cellular origin of myofibroblast. Initially, different works connected the cellular identity for the myofibroblasts origin to resident lung fibroblasts able to directly differentiate into myofibroblasts under profibrotic stimuli [27], epithelial cells undergoing mesenchymal transition, namely, epithelial–mesenchymal transition (EMT) [28][29], bone marrow (BM)-derived cells as circulating fibrocytes [30] and pericytes [31][32]. Lately, the contribution of the epithelial cells through the EMT and the circulating fibrocytes have been objects of several debates [33][34] to such an extent that they are not considered to be cells giving rise to myofibroblasts. To date, given the results obtained from cell-lineage tracing experiments using reporter mouse models, four different cellular types have been demonstrated to acquire the myofibroblastic phenotype [35]. Among them, we found the following: interstitial lung fibroblasts localized in the interstitium immediately adjacent to alveolar epithelial cells [33], lipofibroblasts located near the AECs are lipid-droplet-containing interstitial fibroblasts [35], pericytes within the capillary basement membrane [36], mesothelial cells from pleural-mesothelium that line the visceral and parietal pleural surfaces [37], and resident lung mesenchymal progenitors, whose contribution in the myofibroblast-differentiating process seems prevalent [38]. Since it has been demonstrated that non-IPF ILD may display a progressive phenotype as IPF, opening the possibility to explore the use of antifibrotic drugs for the non-IPF patients, and given the positive outcomes from this, we could speculate that different ILDs may share a similar molecular mechanism that culminates in the fibrogenic phenotype [39][40]. Indeed, since the interstitial lung disease can include rare, heterogeneous and poorly understood diseases, their classification, the identification of the different subtypes, an accurate diagnosis and the prediction of disease progression could be challenging for the clinicians [41].
2. Diagnosis and Histopathological Pattern of IIPs
From a diagnostic point of view, IPF is histologically identified by as usual interstitial pneumonia pattern (UIP) [42], characterized by a heterogenous phenotype with the presence of fibroblastic foci areas interspersed both with an area where the parenchymal lung is mostly preserved and with an area of inflammation and honeycombing. The High-resolution computed tomography (HCRT) of IPF patients shows a peripheral, subpleural and basal pattern of distribution characterized by reticular opacities, honeycombing, minimal ground-glass opacity and architectural distortion [17]. Indeed, the updated version of the American Thoracic Society/European Respiratory Society (ATS/ERS) [19] in 2018 stated an additional recommendation for a multidisciplinary discussion for diagnostic decision making. It occurs that in some cases of interstitial lung disease where there is clinical suspicion of IPF, the HCRT pattern might suggest an alternative diagnosis. Among such cases, nonspecific interstitial pneumonia (NSIP) represents the second most common type of IIP after IPF, accounting for 25% of IIP cases [43]. Furthermore, non-specific interstitial pneumonia (NSIP) can be either idiopathic or associated with connective tissue diseases (e.g., scleroderma), hypersensitivity pneumonia, drug reactions, or diffuse alveolar damage (DAD) and affects more often women ranging from 40 to 50 years of age [23]. The histological pattern of NSIP is characterized by the homogeneous inflammation and expansion of alveolar walls; there are three different subtypes depending on the pattern distribution—cellular, fibrotic and mixed—that are related to different prognoses, where the fibrotic is the representative for the worst prognosis. The HCRT of NSIP patients shows both similarity with IPF patients and peculiar findings that allow for discrimination from radiologists such as extensive ground-glass opacity and the presence of traction bronchiectasis with subpleural sparing in fibrotic non-specific interstitial pneumonia [19]. Indeed, cryptogenic organizing pneumonia (COP), also known as bronchiolitis obliterans organizing pneumonia (BOOP), is mostly found as secondary to connective tissue diseases, drug-induced adverse pulmonary reactions, hypersensitivity pneumonia and infectious processes. It occurs in patients between 50 and 70 years of age, without gender preference, and from the histological point of view it is characterized by organized buds of granulation tissue within the alveolar ducts and adjacent alveoli that create the obstruction of the alveolar lumen and bronchioles culminating in respiratory failure [21][42]. Acute interstitial pneumonia (AIP) is a severe form of idiopathic acute interstitial disease affecting patients between 50 and 60 years of age associated to a poor prognosis with a mortality rate greater than 50%. Although the etiology is unknown, a case report was published that stated how acute interstitial pneumonia can be triggered by strenuous exercise [44]. In general, the clinical, radiological, and histological patterns are similar to those of acute respiratory distress syndrome (ARDS). Typically, the histopathological pattern of AIP is characterized by diffuse alveolar damage (DAD) similar to the pattern found in ARDS [1] with the presence of the edema in the interstitium and alveolus during the early phase of lung injury, followed by the fibroblastic proliferation and type 2 cell hyperplasia in the organizing or late phase [45]. The HRCT shows ground-glass opacities and air space consolidation together with traction bronchiectasis indicating progression from the exudative phase to the proliferative fibrotic phase showing a strong correlation with the different phases of DAD [45]. Desquamative interstitial pneumonia (DIP), which is also a rare pathology mostly affecting men between 30 and 50 years, is strongly related to cigarette smoking and in some case is associated either with occupational risk exposure or the use of inhalation drugs. From a histopathological point of view, DIP is characterized by the thickening of the alveolar septa and massive alveolar infiltration of macrophages leading to interstitial inflammation and fibrosis. The HCRT shows bilateral ground-glass opacities with lower lobe predominance [46]. Lymphocytic interstitial pneumonia (LIP) that is part of the spectrum of benign pulmonary lymphoproliferative disease is characterized by the presence of the lymphocytic infiltrate that expand and culminate into the alveolar region. Furthermore, LIP has been associated with several autoimmune disorders (e.g., Sjögren’s syndrome, rheumatoid arthritis, systemic lupus erythematosus) [47], other pathological conditions such as dysgammaglobulinemia, infections (human immunodeficiency virus and Epstein–Barr virus), and genetic predisposition. The HCRT displays thin-walled cysts in a random distribution, along with ground-glass opacities, centrilobular and subpleural nodules and reticulonodular opacities [18]. Indeed, respiratory bronchiolitis-associated interstitial lung disease (RB-ILD) is a rare, inflammatory pulmonary disorder that mostly affects smokers between 30 and 60 years of age without gender preference. Similar to DIP, RB-ILDs are characterized by the presence of pigmented macrophages within the lumens of respiratory bronchioles and alveolar ducts. The HRCT of RB-ILD patients reveals central and peripheral bronchial wall thickening, centrilobular nodules, and ground-glass opacities together in some case with centrilobular emphysema at the upper lobe. Idiopathic pleuroparenchymal fibroelastosis (PPFE) that has been recently included in the ATS/ERS among the IIPs classification is characterized by the presence of fibrosis in the pleural surfaces and subpleural parenchymal lung, particularly located at the upper lobe. [48] Furthermore, PPFE that mostly occurs in males and with unknown cause [49] has also been reported as a consequence of bone marrow transplantation as chronic graft-versus-host disease [50]. Then, the HRCT of PPFE patients shows pleural thickening in the upper pulmonary zones together with subpleural reticulations in the upper and middle regions and bronchiectasis [49]. Finally, the diagnostic approach through the evaluation of HCRT offers a crucial tool for the diagnosis of IIPs since some radiological patterns specifically and particularly characterize different IIPs (Figure 1). Despite this, sometimes the correct and early diagnosis based on HCRT findings is challenging, especially when the IIPs are represented by heterogeneous patterns shared among/overlapped with other different IIPs as they are for the heterogeneous radiological findings of NSIP. To date, the gold standard for the diagnosis of IIPs, as is the case for the other ILDs, is represented by the multidisciplinary approach that can improve the pathology management and promote an appropriate therapeutic treatment. Indeed, along with the multidisciplinary approach, a more comprehensive study of the molecular mechanisms underlying the specific IIPs as well as of the shared molecular mechanisms could support both the diagnosis and the prognosis of these patients as well as widen the perspectives for potential new target therapies.
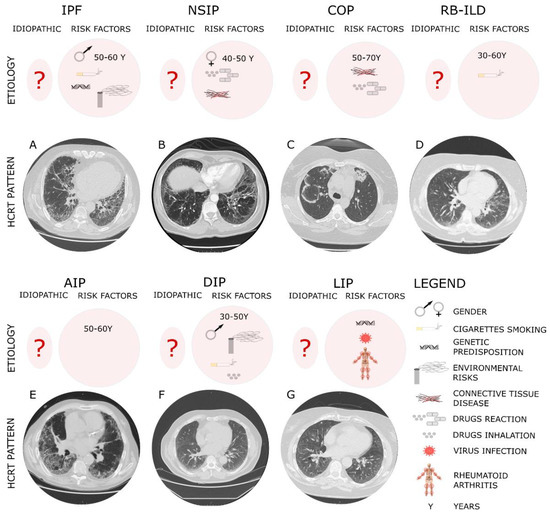
Figure 1. The diagnostic and etiologic characteristic of different IIPs.
The typical high-resolution computed tomography (HRCT) of different IIPs shows the UIP pattern where the main features are represented by a reticular pattern, traction bronchiectasis and honeycombing appearance. These imaging findings are predominantly located in the subpleural regions and in lower lobes. In particular, IPF, which is considered idiopathic (?) but has several risk factors, such as the gender and age, genetic susceptibility, environmental risks and cigarette smoking, shows in Panel A peripheral, subpleural and basal pattern of distribution characterized by reticular opacities, honeycombing, minimal ground-glass opacity and architectural distortion. NSIP that can be either idiopathic or associated with connective tissue diseases (e.g., scleroderma), hypersensitivity pneumonia and drug reactions, more often affecting women ranging from 40 to 50 years of age, shows (Panel B) from the HCRT extensive ground-glass opacity and the presence of traction bronchiectasis with subpleural sparing in fibrotic non-specific interstitial pneumonia. COP, mostly found as secondary to connective tissue diseases, drug-induced adverse pulmonary reactions, hypersensitivity pneumonia and infectious processes affecting patients between 50 and 70 years of age, shows a radiological pattern (Panel C) characterized by peripheral or peribronchial patchy consolidations as well as ground-glass opacities with a tendency to migrate. RB-ILD mostly affects smokers between 30 and 60 years of age without gender preference. The HRCT of RB-ILD patients (Panel D) reveals central and peripheral bronchial wall thickening, centrilobular nodules, and ground-glass opacities together in some case with centrilobular emphysema at the upper lobe. AIP affects patients between 50 and 60 years of age with unknown etiology. The HRCT (Panel E) shows ground-glass opacities and air space consolidation together with traction bronchiectasis. DIP, mostly affecting men between 30 and 50 years, is strongly related to cigarette smoking, occupational risk exposure or use of inhalation drugs. The HCRT (Panel F) shows bilateral ground-glass opacities with lower lobe predominance. LIP has been associated with several autoimmune disorders (e.g., Sjögren’s syndrome, rheumatoid arthritis, systemic lupus erythematosus), virus and genetic predisposition. The HCRT (Panel G) displays thin-walled cysts in a random distribution, along with ground-glass opacities, centrilobular and subpleural nodules and reticulonodular opacities.
References
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E.; Lynch, D.A.; Nicholson, A.G.; Wells, A.U.; Behr, J.; Bouros, D.; Ryerson, C.J.; et al. American Thoracic Society Documents An Official American Thoracic Society/European Respiratory Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748.
- Wong, A.W.; Ryerson, C.J.; Guler, S.A. Progression of fibrosing interstitial lung disease. Respir. Res. 2020, 21, 1–10.
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952.
- Jee, A.S.; Corte, T.J. Current and Emerging Drug Therapies for Connective Tissue Disease-Interstitial Lung Disease (CTD-ILD). Drugs 2019, 79, 1511–1528.
- Wells, A.U.; Denton, C.P. Interstitial lung disease in connective tissue disease-Mechanisms and management. Nat. Rev. Rheumatol. 2014, 10, 728–739.
- Rivera-Ortega, P.; Molina-Molina, M. Interstitial lung diseases in developing countries. Ann. Glob. Health 2019, 85, 1–14.
- Gulati, M.; Redlich, C.A. Asbestosis and environmental causes of usual interstitial pneumonia. Curr. Opin. Pulm. Med. 2015, 21, 193–200.
- Barber, C.M.; Sherwood Burge, P.; Feary, J.R.; Parfrey, H.; Renzoni, E.A.; Spencer, L.G.; Walters, G.I.; Wiggans, R.E. Identifying causation in hypersensitivity pneumonitis: A British perspective. BMJ Open Respir. Res. 2019, 6, 1–6.
- Ryerson, C.J.; Vittinghoff, E.; Ley, B.; Lee, J.S.; Mooney, J.J.; Jones, K.D.; Elicker, B.M.; Wolters, P.J.; Koth, L.L.; King, T.E.; et al. Predicting survival across chronic interstitial lung disease: The ILD-GAP model. Chest 2014, 145, 723–728.
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824.
- Collard, H.R.; Ryerson, C.J.; Corte, T.J.; Jenkins, G.; Kondoh, Y.; Lederer, D.J.; Lee, J.S.; Maher, T.M.; Wells, A.U.; Antoniou, K.M.; et al. Acute exacerbation of idiopathic pulmonary fibrosis an international working group report. Am. J. Respir. Crit. Care Med. 2016, 194, 265–275.
- Meyer, K.C. Pulmonary fibrosis, part I: Epidemiology, pathogenesis, and diagnosis. Expert Rev. Respir. Med. 2017, 11, 343–359.
- Cerri, S.; Monari, M.; Guerrieri, A.; Donatelli, P.; Bassi, I.; Garuti, M.; Luppi, F.; Betti, S.; Bandelli, G.; Carpano, M.; et al. Real-life comparison of pirfenidone and nintedanib in patients with idiopathic pulmonary fibrosis: A 24-month assessment. Respir. Med. 2019, 159, 105803.
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082.
- Guenther, A.; Krauss, E.; Tello, S.; Wagner, J.; Paul, B.; Kuhn, S.; Maurer, O.; Heinemann, S.; Costabel, U.; Barbero, M.A.N.; et al. The European IPF registry (eurIPFreg): Baseline characteristics and survival of patients with idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 1–10.
- Belloli, E.A.; Beckford, R.; Hadley, R.; Flaherty, K.R. Idiopathic non-specific interstitial pneumonia. Respirology 2016, 21, 259–268.
- Oliveira, D.; de Arimatéia Araújo Filho, J.; Fernando Lins Paiva, A.; Seigo Ikari, E.; Caruso Chate, R.; Higa Nomura, C. Idiopathic interstitial pneumonias: Review of the latest American Thoracic Society/European Respiratory Society classification. Radiol. Bras. 2018, 51, 321–327.
- Panchabhai, T.S.; Farver, C.; Highland, K.B. Lymphocytic Interstitial Pneumonia. Clin. Chest Med. 2016, 37, 463–474.
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of idiopathic pulmonary fibrosis An Official ATS/ERS/JRS/ALAT Clinical practice guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68.
- Sverzellati, N.; Lynch, D.A.; Hansell, D.M.; Johkoh, T.; King, T.E.; Travis, W.D. American Thoracic Society-European Respiratory Society Classification of the Idiopathic Interstitial Pneumonias: Advances in Knowledge since 2002. Radiographics 2015, 35, 1849–1872.
- Guidotti, T.L.; Miller, A.; Christiani, D.; Wagner, G.; Balmes, J.; Harber, P.; Brodkin, C.A.; Rom, W.; Hillerdal, G.; Harbut, M.; et al. Diagnosis and initial management of nonmalignant diseases related to asbestos. Am. J. Respir. Crit. Care Med. 2004, 170, 691–715.
- Leung, C.C.; Yu, I.T.S.; Chen, W. Silicosis. Lancet 2012, 379, 2008–2018.
- Ebner, L.; Christodoulidis, S.; Stathopoulou, T.; Geiser, T.; Stalder, O.; Limacher, A.; Heverhagen, J.T.; Mougiakakou, S.G.; Christe, A. Meta-analysis of the radiological and clinical features of Usual Interstitial Pneumonia (UIP) and Nonspecific Interstitial Pneumonia (NSIP). PLoS ONE 2020, 15, e0226084.
- Selman, M.; Pardo, A. The leading role of epithelial cells in the pathogenesis of idiopathic pulmonary fibrosis. Cell. Signal. 2020, 66, 109482.
- Pardo, A.; Selman, M. The Interplay of the Genetic Architecture, Aging, and Environmental Factors in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 64, 163–172.
- Hao, Y.; Bates, S.; Mou, H.; Yun, J.H.; Pham, B.; Liu, J.; Qiu, W.; Guo, F.; Morrow, J.D.; Hersh, C.P.; et al. Genome-wide association study: Functional variant rs2076295 regulates desmoplakin expression in airway epithelial cells. Am. J. Respir. Crit. Care Med. 2020, 202, 1225–1236.
- Phan, S.H. The myofibroblast in pulmonary fibrosis. Chest 2002, 122, 286S–289S.
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; Du Bois, R.M.; Borok, Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-β1: Potential role in idiopathic pulmonary fibrosis. Am. J. Pathol. 2005, 166, 1321–1332.
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.M.; Sheppard, D.; Chapman, H.A. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185.
- Phillips, R.J.; Burdick, M.D.; Hong, K.; Lutz, M.A.; Murray, L.A.; Xue, Y.Y.; Belperio, J.A.; Keane, M.P.; Strieter, R.M. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J. Clin. Investig. 2004, 114, 438–446.
- Hung, C.; Linn, G.; Chow, Y.; Kobayashi, A.; Mittelsteadt, K.; Altemeier, W.A.; Gharib, S.A.; Schnapp, L.M.; Duffield, J.S. Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2013, 188, 820–830.
- Hinz, B. Mechanical aspects of lung fibrosis: A spotlight on the myofibroblast. Proc. Am. Thorac. Soc. 2012, 9, 137–147.
- Barron, L.; Gharib, S.A.; Duffield, J.S. Lung Pericytes and Resident Fibroblasts: Busy Multitaskers. Am. J. Pathol. 2016, 186, 2519–2531.
- Chong, S.G.; Sato, S.; Kolb, M.; Gauldie, J. Fibrocytes and fibroblasts—Where are we now. Int. J. Biochem. Cell Biol. 2019, 116, 105595.
- Al Alam, D.; El Agha, E.; Sakurai, R.; Kheirollahi, V.; Moiseenko, A.; Danopoulos, S.; Shrestha, A.; Schmoldt, C.; Quantius, J.; Herold, S.; et al. Evidence for the involvement of fibroblast growth factor 10 in lipofibroblast formation during embryonic lung Development. Development 2015, 142, 4139–4150.
- Marriott, S.; Baskir, R.S.; Gaskill, C.; Menon, S.; Carrier, E.J.; Williams, J.; Talati, M.; Helm, K.; Alford, C.E.; Kropski, J.A.; et al. ABCG2pos lung mesenchymal stem cells are a novel pericyte subpopulation that contributes to fibrotic remodeling. Am. J. Physiol. Cell Physiol. 2014, 307, C684–C698.
- Karki, S.; Surolia, R.; Hock, T.D.; Guroji, P.; Zolak, J.S.; Duggal, R.; Ye, T.; Thannickal, V.J.; Antony, V.B. Wilms’ tumor 1 (Wt1) regulates pleural mesothelial cell plasticity and transition into myofibroblasts in idiopathic pulmonary fibrosis. FASEB J. 2014, 28, 1122–1131.
- Xie, T.; Liang, J.; Liu, N.; Huan, C.; Zhang, Y.; Liu, W.; Kumar, M.; Xiao, R.; D’Armiento, J.; Metzger, D.; et al. Transcription factor TBX4 regulates myofibroblast accumulation and lung fibrosis. J. Clin. Investig. 2016, 126, 3063–3079.
- Wells, A.U.; Flaherty, K.R.; Brown, K.K.; Inoue, Y.; Devaraj, A.; Richeldi, L.; Moua, T.; Crestani, B.; Wuyts, W.A.; Stowasser, S.; et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases—subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: A randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir. Med. 2020, 8, 453–460.
- Maher, T.M.; Corte, T.J.; Fischer, A.; Kreuter, M.; Lederer, D.J.; Molina-Molina, M.; Axmann, J.; Kirchgaessler, K.U.; Samara, K.; Gilberg, F.; et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir. Med. 2020, 8, 147–157.
- Adegunsoye, A.; Ryerson, C.J. Diagnostic Classification of Interstitial Lung Disease in Clinical Practice. Clin. Chest Med. 2021, 42, 251–261.
- Lynch, D.A.; Travis, W.D.; Müller, N.L.; Galvin, J.R.; Hansell, D.M.; Grenier, P.A.; King, T.E. Idiopathic interstitial pneumonias: CT features. Radiology 2005, 236, 10–21.
- Travis, W.D.; Hunninghake, G.; King, T.E.; Lynch, D.A.; Colby, T.V.; Galvin, J.R.; Brown, K.K.; Man, P.C.; Cordier, J.F.; Du Bois, R.M.; et al. Idiopathic nonspecific interstitial pneumonia: Report of an American Thoracic Society Project. Am. J. Respir. Crit. Care Med. 2008, 177, 1338–1347.
- Markoska, F.; Lestan, D.; Turel, M.; Harlander, M. Respiratory Medicine Case Reports Acute interstitial pneumonia triggered by strenuous exercise. Respir. Med. Case Rep. 2020, 30, 101077.
- Ichikado, K. Syndrome, Acute Interstitial Pneumonia. Semin. Ultrasound CT MRI 2014, 35, 39–46.
- Hellemons, M.E.; Moor, C.C.; Von Der Thüsen, J.; Rossius, M.; Odink, A.; Thorgersen, L.H. Desquamative interstitial pneumonia: A systematic review of its features and outcomes. Eur. Respir. Rev. 2020, 29, 190181.
- Luppi, F.; Sebastiani, M.; Silva, M.; Sverzellati, N.; Cavazza, A.; Salvarani, C.; Manfredi, A.; Salvarani, C. Review Interstitial lung disease in Sjögren ’ s syndrome: A clinical review. Clin. Exp. Rheumatol. 2020, 38, 291–300.
- Palmucci, S.; Roccasalva, F.; Puglisi, S.; Torrisi, S.E.; Vindigni, V.; Mauro, L.A.; Ettorre, G.C.; Piccoli, M. Clinical and radiological features of idiopathic interstitial pneumonias (IIPs): A pictorial review. Insights Imaging 2014, 5, 347–364.
- Kusagaya, H.; Nakamura, Y.; Kono, M.; Kaida, Y.; Kuroishi, S.; Enomoto, N.; Fujisawa, T.; Koshimizu, N.; Yokomura, K.; Inui, N.; et al. Idiopathic pleuroparenchymal fibroelastosis: Consideration of a clinicopathological entity in a series of Japanese patients. BMC Pulm. Med. 2012, 12, 72.
- Fomby, P.; Cherlin, A.J.; Hadjizadeh, A.; Doillon, C.J.; Sueblinvong, V.; Weiss, D.J.; Bates, J.H.T.; Gilbert, T.; Liles, W.C.; Lutzko, C.; et al. Stem cells and cell therapies in lung biology and diseases: Conference report. Ann. Am. Thorac. Soc. 2010, 12, 181–204.
More
Information
Subjects:
Respiratory System
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
890
Revisions:
2 times
(View History)
Update Date:
31 Aug 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No