Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lehang Lin | + 2722 word(s) | 2722 | 2021-08-18 11:43:41 | | | |
| 2 | Vivi Li | Meta information modification | 2722 | 2021-08-31 08:13:19 | | | | |
| 3 | Conner Chen | Meta information modification | 2722 | 2021-10-12 10:55:57 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lin, L. Tumor Heterogeneity in ESCC. Encyclopedia. Available online: https://encyclopedia.pub/entry/13718 (accessed on 07 February 2026).
Lin L. Tumor Heterogeneity in ESCC. Encyclopedia. Available at: https://encyclopedia.pub/entry/13718. Accessed February 07, 2026.
Lin, Lehang. "Tumor Heterogeneity in ESCC" Encyclopedia, https://encyclopedia.pub/entry/13718 (accessed February 07, 2026).
Lin, L. (2021, August 30). Tumor Heterogeneity in ESCC. In Encyclopedia. https://encyclopedia.pub/entry/13718
Lin, Lehang. "Tumor Heterogeneity in ESCC." Encyclopedia. Web. 30 August, 2021.
Copy Citation
Esophageal squamous cell carcinoma (ESCC) is a common and aggressive malignancy, with hitherto dismal clinical outcome. Genomic analyses of patient samples reveal a complex heterogeneous landscape for ESCC, which presents in both intertumor and intratumor forms, manifests at both genomic and epigenomic levels, and contributes significantly to tumor evolution, drug resistance, and metastasis.
esophageal squamous cell carcinoma
tumor heterogeneity
tumor evolution
precision medicine
1. Introduction
Esophageal carcinoma is the sixth most lethal cancer type worldwide, responsible for over 400,000 deaths annually [1][2]. Esophageal squamous cell carcinoma (ESCC) is the predominant histological subtype, accounting for 90% of cases [3][4]. Despite noteworthy advances in both cancer diagnosis and therapy, the clinical outlook for ESCC patients remains dismal, with a five-year survival rate below 30% [5][6]. A number of lines of evidence have demonstrated that this poor clinical outcome is at least partially attributed to the substantial intertumor and intratumor heterogeneity in ESCC [7][8].
The concept of tumor heterogeneity contains both intertumor and intratumor forms. Intertumor heterogeneity concerns the phenotypic and molecular differences among tumors from different patients, while intratumor heterogeneity refers to biological variations within the same tumor [9][10][11][12][13]. Heterogeneity is an important attribute of cancer and a major contributor to tumor progression. It manifests at two major levels: genomic (somatic mutations, copy number alterations, chromosomal rearrangements, etc.) and non-genomic (epigenomic changes, microenvironmental variabilities, etc.) [14][15]. The degree and complexity of tumor heterogeneity influence the strategy of tumor biopsy, cancer diagnosis, and treatment planning [7][9][14][16][17][18]. Increasingly, advances in sequencing technology and analysis algorithms have substantially promoted the understanding of both intertumor and intratumor heterogeneity in many cancer types, including ESCC [7][19][20]. However, translation of the accumulated knowledge on ESCC heterogeneity into clinical practice is still challenging. A systematic understanding of ESCC heterogeneity with respect to its composition, function, and implication is therefore urgently needed.
2. Intertumor Heterogeneity
Taxonomy of cancer subtypes by specific molecular characteristics significantly improves the conventional histopathological classification and guides subtype-specific precision medicine. As exemplified in breast cancer (e.g., luminal, basal-like, Her2+), lung cancer (e.g., EGFR+, ALK fusion+), and gastric cancer (e.g., Epstein–Barr virus+, microsatellite unstable), intertumor heterogeneity has been widely studied and successfully translated into clinical knowledge in various cancer types [21][22][23]. However, the stratification of ESCC patients based on intertumoral molecular heterogeneity remains comparatively understudied.
In 2017, through an integrative multi-omics analysis, The Cancer Genome Atlas (TCGA) consortium classified 90 ESCC specimens into three subtypes, designated as ESCC1–3 [4]. ESCC1, mostly Asian samples, was enriched in genomic alterations in the NRF2 pathway (NFE2L2, KEAP1, CUL3, and ATG7) and amplifications of SOX2 and/or TP63; ESCC2, mainly Eastern European and South American samples, was characterized by higher rates of NOTCH1 and ZNF750 mutations, CDK6 amplification, and inactivation of KDM6A, KDM2D, PTEN, and PIK3R1; only four cases were classified into ESCC3, which were all from North America and featured in SMARCA4 mutation. Although these subtypes showed notable geographical trends, their associations with particular biological and/or clinical features were not extensively elucidated. In addition, because of the relatively small number of samples, these classifications need further validation in larger cohorts.
In addition to the effort from TCGA consortium, several individual laboratories have attempted to subgroup ESCC based on transcriptomic data. Upon analyzing tumor samples from African patients, Liu et al. [24] reported three ESCC subtypes based on their distinct expression patterns of cell cycle and neural transcripts. In another study, ESCC specimens from 360 East Asian individuals were divided into four molecular subtypes associated with distinct clinical metrics [25]. Most recently, a new research work has categorized Asian ESCCs into two subtypes, with subtype I overexpressing genes in immune response process and subtype II linked to ectoderm development, cell proliferation, and glycolysis process [26]. Additionally, Tanaka et al. [27] reported the presence of an immune-reactive subtype of ESCC patients with cytotoxic T-lymphocyte signatures activated by chemoradiotherapy.
Despite the fact that no consensus molecular subtypes of ESCC have been established, the above subtyping results are sufficient to confirm the existence of extensive intertumor heterogeneity among ESCC individuals, and further demonstrate heterogeneity amongst different ESCC ethnic groups. This is in line with the well-established dramatic geographic and demographic features of ESCC [28][29]. It should also be noted that the molecular factors and causes underlying intertumor heterogeneity are likely similar with those involved in intratumor diversity. In order to fully capture the tumor spectrum, and to further improve ESCC subclassification and treatment stratification, the molecular features of ESCC intratumor heterogeneity need to be comprehensively integrated.
3. Intratumor Heterogeneity
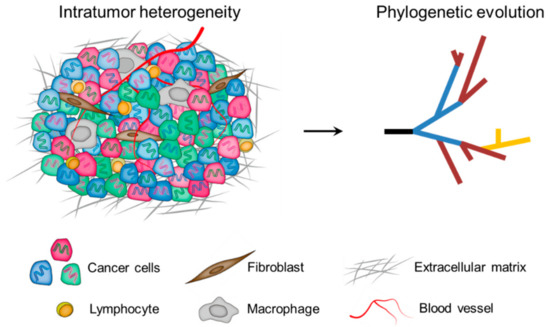
In the milestone paper published in 1976, Peter C. Nowell [30] proposed a model for cancer development: the Darwinian clonal evolution and selection of tumor cells. Since then, this model has been widely accepted and the phenomenon of intratumor heterogeneity has been highlighted as a cancer hallmark to reflect the non-uniformity and intricacy within tumor ecosystems [31][32][33]. To date, it is well established that intratumor heterogeneity is represented by the presence of distinct cell populations, which can occupy specific microenvironmental niches, behave as communities, and extensively interact with each other as well as with components of the tumor microenvironment [12]. Therefore, intratumor heterogeneity arises not only from genomic and epigenomic disorders of tumor cells themselves, but also from the influence of the tumor microenvironment [9]. Importantly, intratumor heterogeneity exists among different geographical regions of the same tumor (spatial heterogeneity), as well as between the primary tumor and subsequent local or distant recurrence in the same patient (temporal heterogeneity). As a cumulative result, tumor cells display remarkable variability in numerous phenotypic traits, including clinically important phenotypes such as the ability to seed metastases and to survive therapy (Figure 1) [34].
Figure 1. Multiple layers of intratumor heterogeneity. Left: Phenotypically distinct cancer cells with both genomic (DNA color) and epigenomic (cell color) heterogeneity are admixed with diversified microenvironmental components. Right: A phylogenetic framework helps to understand the nature and biological significance of tumor spatiotemporal heterogeneity.
3.1. Clonal Evolution of Tumors
According to the clonal evolution hypothesis, cancer arises from a single founder cell, and tumor progression is accompanied by the resultant succession of clonal expansions that follow the Darwinian logic [30]. This evolutionary perspective underlines genomic alterations as an essential substrate for fueling tumor transformation and evolution.
During each cell cycle, regardless of normal or cancer cells, DNA mutations may be acquired. Thus, the acquisition of mutations is a stochastic and random process. Consequently, innumerable rounds of cell divisions required for the formation of macroscopic tumors offer plenty of opportunities for Darwinian selection and emergence of clonal diversity in tumor cell populations. During clonal evolution, only a few “jackpot” mutations that activate oncogenic pathways and/or inactivate tumor suppressors are selectively advantageous, allowing the mutant clones to achieve selective sweeps. These functionally significant mutations are termed “drivers”. In contrast, the vast majority of mutations are functionally neutral since they do not confer competitive fitness advantage. These mutations are so-called “passengers” and are mainly responsible for intratumor heterogeneity [35]. Importantly, clonal evolution often proceeds in a branching rather than in a linear manner, further contributing to variegated tumor subclones and the complexity of tumor evolution [14]. In fact, many neutral or mildly deleterious mutations during clonal expansion can be retained in the population, or even undergo expansions due to the genetic drift [32][35]. Moreover, given the fact that the Darwinian selection is context-specific, and the evolutionary dynamics of tumor microenvironment and epigenomic events could translate into heterogeneous selective pressures experienced by tumor cells, the selective effect of given mutations (either driver or passenger) can change substantially at different stages of tumor progression [14].
3.2. Spatial Intratumor Heterogeneity
Spatial intratumor heterogeneity has been elucidated at high resolution in many cancer types [15][36][37][38][39][40]. Recently, several groups have performed multi-regional deep-sequencing, and have presented a comprehensive heterogeneous landscape of ESCC [41][42][43][44]. Through analyzing 51 sub-tumor regions from 13 ESCC patients, Hao et al. [42] proposed that approximately 40% of driver mutations were spatially heterogeneous, including oncogenes such as KIT, and members of the PI3K/MTOR (PIK3CA and MTOR) and NFE2L2 pathways (NFE2L2 and KEAP1). In addition, significant spatial heterogeneity was observed in copy number alterations, including EGFR amplification and CDKN2A/B deletions [42]. Furthermore, taking into consideration the multi-step progression of ESCC, Zeng’s team [43] sequenced different segments of ESCC tumors and their matched dysplasia samples in a cohort of 20 patients. Their analyses showed that esophagus dysplasia also carried high mutation load and, remarkably, more heterogeneous mutations were seen in dysplasia than in tumor samples from each patient. Moreover, through sequencing 682 micro-scale esophageal samples, Yokoyama et al. [45] reported very recently that pervasive expansions of multiple independent clones were more commonly present within physiologically normal esophagus in comparison to ESCCs. These seemingly surprising data indicate that diversified mutational backgrounds were already established in the precursor lesion or even normal esophageal epithelia, conferring on the esophageal cells the ability to evade selection pressure during ESCC development. Moreover, the degree and complexity of spatial heterogeneity was found to be highly correlated with ESCC aggressiveness [44]. Specifically, clinical stage of ESCC was negatively correlated with the proportion of ubiquitous mutations, and significantly more heterogeneous mutations were observed in ESCC patients with local metastasis, compared to those without.
Regionally segregated somatic mutations and copy number alterations have important clinical implications in ESCC. Firstly, they complicate pathological evaluation of tumor samples. Owing to potential sampling bias caused by spatial heterogeneity, the representability of tumor regions subject to pathological assessment is increasingly considered as a key factor. It is possible that diagnostic and therapeutic targets located in uninspected regions are missed by chance, and the heterogeneous spectrum of the tumor is inevitably underestimated. Additionally, spatial genomic heterogeneity is an important determinant for therapeutic responses. Although most cancers initially respond to treatment, they almost always relapse with the outgrowth of cancer cells that are no longer sensitive to the therapy. Many cases have demonstrated that resistance to targeted drugs may result from the preexisting heterogeneous cells. Examples include the impaired efficiency of EGFR inhibitor for lung cancer patients with heterogeneous driver status [46][47]. Lung cancers initially containing rare mutations of EGFR, e.g., T790M, or low frequency of MET amplification, are capable of rendering resistance to targeted therapy [18][31][48][49]. Another well-understood case is chronic myeloid leukemia, in which mutant forms of the BCR-ABL fusion protein have been implicated in the relapse of disease under imatinib treatment [50][51][52]. In ESCC, heterogeneous amplifications of EGFR, FGFR1, and PD-L1 have been reported [42][43][44], accounting partially for the unsatisfactory efficacy of targeting such genomic lesions [53][54][55]. Spatial genomic heterogeneity, therefore, greatly challenges both accurate diagnosis and efficient cancer treatment.
In addition to genomic alterations, epigenomic dysregulation also contributes to spatial diversity within a tumor. Mechanistically, epigenomic heterogeneity may arise from changes in chromatin status (e.g., DNA methylation, histone modification), deregulation of microRNAs, and transcription regulators, etc. These alterations potentially provide fitness benefit, leading to intratumor heterogeneity either independently or in conjunction with genomic alterations [56][57][58][59][60][61][62][63][64]. For example, DNA methylation status within promoters of transcription factors SIM2 and SIX1 was strongly correlated with their heterogeneous expression pattern, which was further associated with ESCC differentiation, progression, and prognosis [65][66][67]. Dynamic changes of mutational status and promoter DNA methylation were also observed in the SWI/SNF chromatin remodeling complex and were shown to involve in ESCC carcinogenesis [68]. Moreover, epigenomic and genomic heterogeneity have been integratively analyzed in three ESCC patients [42]. Noticeably, the spatial heterogeneous pattern of DNA methylation closely recapitulated that of somatic mutations, indicating functional interplay between genomic and epigenomic alterations in ESCC.
The tumor microenvironment, consisting of fibroblasts, extracellular matrix, immune cells (e.g., macrophages, infiltrating lymphocytes), etc., imposes yet another layer of heterogeneity [44][69][70][71][72][73][74][75]. Tumor microenvironment can shape tumor cell phenotypes by augmenting both the intrinsic variability of cancer cells (e.g., by inducing stress responses and genomic instability) and the extrinsic diversity of microenvironmental contexts (e.g., different densities of blood and lymphatic vasculature, different numbers and types of infiltrating cells) [7]. In ESCC, the tumor microenvironment itself is indeed highly heterogeneous, as evidenced by recent reports of intratumor heterogeneity of tumor infiltrating T and B cells [44][70][75]. Additionally, Yan et al. [44] observed a tight association between genomic heterogeneity and variation of T cell repertoire in ESCC primary tumors. These results demonstrate that the intratumor genomic heterogeneity may have clinical relevance in ESCC through affecting tumor microenvironment. Meanwhile, ESCC cells could also benefit from the microenvironmental heterogeneity, which supports cellular diversity and influences evolutionary trajectories [14][62][76][77].
3.3. Temporal Intratumor Heterogeneity
Accumulating evidence suggests that intratumor heterogeneity contributes to tumor growth through a process called branched evolution. This model suggests that tumorigenesis is analogous to a growing tree, whose trunk gives rise to numerous branches [9][14][78]. Phylogenetic analysis is a useful approach to delineate such tree structure of cancer evolution [19][38][79][80][81]. Accordingly, in the phylogenetic tree, truncal (ubiquitous) events shared by the entire tumor population likely reflect processes involved before and during tumor initiation and early development, whereas branched (heterogeneous) events present in only some regions of the tumor reveal factors shaping the genome during tumor maintenance and progression. Characterization of the relative timing of key somatic events with possible biological relevance is therefore essential for deciphering the evolutionary processes of tumors, as well as further improving precision medicine strategies.
In ESCC, driver mutations were significantly more truncal/clonal than passenger mutations, in accordance with findings in other tumor types. Importantly, the majority of driver mutations in tumor suppressors (including TP53, KMT2D, ZNF750, etc.) had a tendency to locate in the trunks of phylogenetic trees, indicating that tumor suppressors are lost as relatively early events during ESCC development. In contrast, half of the driver mutations in the branches were in oncogenes, including potential actionable targets, PIK3CA and MTOR, suggesting that they are late events in ESCC [42]. This observation highlights the extra caution needed when considering inhibiting such oncogenic mutants in ESCC, given previous studies showing that suppressing subclonal drivers could otherwise lead to outgrowth of non-mutated subpopulations [82].
Esophageal squamous cell carcinoma evolution is a multi-step process that begins from low-grade dysplasia, high-grade dysplasia, carcinoma in situ to invasive tumor and metastasis [44]. To further explore the genomic dynamics during this process, recent studies applied multi-region sequencing on samples covering different stages of ESCC from the same patients and constructed phylogenetic trees that mapped mutations and copy number alterations chronologically [43][44][83]. Notably, only a small fraction of total genomic alterations was conserved from squamous dysplasia to ESCC tumors, implying the distinct evolutionary trajectories taken by precursor and neoplastic cells [43][83]. Phylogenetic analysis confirmed truncal mutations of TP53 and CDKN2A and truncal copy number alterations of 11q13 (CCND1), 3q27 (SOX2), 2q31 (NFE2L2), and 9p21 (CDKN2A), validating that they are early changes during esophagus neoplastic transformation [83]. Independently, Chen et al. [43] also reported early emergence of copy number alterations in precursor lesions of ESCC and highlighted this phenomenon as a prominent genomic feature distinct from the development of esophageal adenocarcinoma, another pathological subtype of esophageal cancer. When considering alterations at pathway level, genes involved in cell cycle regulation (such as TP53, CCND1, CDK6, RB1, and CDKN2A) were frequently altered in the early stage of ESCC, whereas genes in RTK/RAS/PI3K tended to undergo alterations throughout the process of ESCC evolution [44].
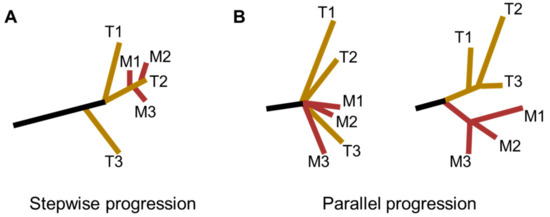
Taking into consideration of the timing of metastatic outgrowth and the role of the intratumor heterogeneity, two distinct models for the derivation of ESCC metastasis have been proposed: the stepwise progression model and the parallel progression model (Figure 2) [43][44]. The stepwise progression model was characterized by tumor cells disseminated at the late stages of ESCC. Accordingly, metastases could be considered as direct descendants of the most malignant and aggressive clones that dominated primary tumors. This model was also described as the linear spread pattern by Yan et al. [44]. By comparison, in the parallel progression model, early spread of metastases during ESCC tumor progression was highlighted. Specifically, divergent evolutionary trajectories were found between primary tumors and metastatic lesions, as well as among metastatic lesions. This model was represented as both explosive spread and metastasis-to-metastasis patterns by Yan and colleagues [44].
Figure 2. Phylogenic models for ESCC metastasis. (A) The stepwise progression model, in which metastases are seeded at the late stages of ESCC progression. This model was also described as the linear spread pattern by Yan et al. [44]. (B) The parallel progression model, in which tumor dissemination occurs at the early stages of ESCC progression. In this situation, dissemination of metastases could happen either (Left) explosively (explosive spread pattern) or via (Right) metastasis-to-metastasis pattern (T, Tumor; M, Metastasis).
More studies are required to elucidate the clonal relationship between ESCC primary and metastatic tumor cell populations, which will not only illuminate the evolutionary history of ESCC, but also create a more solid ground for therapeutic decision making. Ultimately, decoding the extent of differences between ESCC primary and metastases is crucial for the improved management of metastatic ESCC patients.
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108.
- Lin, D.C.; Wang, M.R.; Koeffler, H.P. Genomic and Epigenomic Aberrations in Esophageal Squamous Cell Carcinoma and Implications for Patients. Gastroenterology 2018, 154, 374–389.
- Rustgi, A.K.; El-Serag, H.B. Esophageal carcinoma. N. Engl. J. Med. 2014, 371, 2499–2509.
- Cancer Genome Atlas Research Network. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175.
- Pennathur, A.; Gibson, M.K.; Jobe, B.A.; Luketich, J.D. Oesophageal carcinoma. Lancet 2013, 381, 400–412.
- Napier, K.J.; Scheerer, M.; Misra, S. Esophageal cancer: A Review of epidemiology, pathogenesis, staging workup and treatment modalities. World J. Gastrointest. Oncol. 2014, 6, 112–120.
- Yap, T.A.; Gerlinger, M.; Futreal, P.A.; Pusztai, L.; Swanton, C. Intratumor heterogeneity: Seeing the wood for the trees. Sci. Transl. Med. 2012, 4, 127ps10.
- Gerlinger, M.; Swanton, C. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br. J. Cancer 2010, 103, 1139–1143.
- Gerashchenko, T.S.; Denisov, E.V.; Litviakov, N.V.; Zavyalova, M.V.; Vtorushin, S.V.; Tsyganov, M.M.; Perelmuter, V.M.; Cherdyntseva, N.V. Intratumor heterogeneity: Nature and biological significance. Biochemistry 2013, 78, 1201–1215.
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322.
- Wolman, S.R.; Heppner, G.H. Genetic heterogeneity in breast cancer. J. Natl. Cancer Inst. 1992, 84, 469–470.
- Tabassum, D.P.; Polyak, K. Tumorigenesis: It takes a village. Nat. Rev. Cancer 2015, 15, 473–483.
- Axelrod, R.; Axelrod, D.E.; Pienta, K.J. Evolution of cooperation among tumor cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13474–13479.
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334.
- Navin, N.; Krasnitz, A.; Rodgers, L.; Cook, K.; Meth, J.; Kendall, J.; Riggs, M.; Eberling, Y.; Troge, J.; Grubor, V.; et al. Inferring tumor progression from genomic heterogeneity. Genome Res. 2010, 20, 68–80.
- Swanton, C. Intratumor heterogeneity: Evolution through space and time. Cancer Res. 2012, 72, 4875–4882.
- Horswell, S.; Matthews, N.; Swanton, C. Cancer heterogeneity and “the struggle for existence”: Diagnostic and analytical challenges. Cancer Lett. 2013, 340, 220–226.
- Fisher, R.; Pusztai, L.; Swanton, C. Cancer heterogeneity: Implications for targeted therapeutics. Br. J. Cancer 2013, 108, 479–485.
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892.
- Van Nistelrooij, A.M.; van Marion, R.; Koppert, L.B.; Biermann, K.; Spaander, M.C.; Tilanus, H.W.; van Lanschot, J.J.; Wijnhoven, B.P.; Dinjens, W.N. Molecular clonality analysis of esophageal adenocarcinoma by multiregion sequencing of tumor samples. BMC Res. Notes 2017, 10, 144.
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209.
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550.
- Heiser, L.M.; Sadanandam, A.; Kuo, W.L.; Benz, S.C.; Goldstein, T.C.; Ng, S.; Gibb, W.J.; Wang, N.J.; Ziyad, S.; Tong, F.; et al. Subtype and pathway specific responses to anticancer compounds in breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 2724–2729.
- Liu, W.; Snell, J.M.; Jeck, W.R.; Hoadley, K.A.; Wilkerson, M.D.; Parker, J.S.; Patel, N.; Mlombe, Y.B.; Mulima, G.; Liomba, N.G.; et al. Subtyping sub-Saharan esophageal squamous cell carcinoma by comprehensive molecular analysis. JCI Insight 2016, 1, e88755.
- Xiong, T.; Wang, M.; Zhao, J.; Liu, Q.; Yang, C.; Luo, W.; Li, X.; Yang, H.; Kristiansen, K.; Roy, B.; et al. An esophageal squamous cell carcinoma classification system that reveals potential targets for therapy. Oncotarget 2017, 8, 49851–49860.
- Wang, F.; Yan, Z.; Lv, J.; Xin, J.; Dang, Y.; Sun, X.; An, Y.; Qi, Y.; Jiang, Q.; Zhu, W.; et al. Gene Expression Profiling Reveals Distinct Molecular Subtypes of Esophageal Squamous Cell Carcinoma in Asian Populations. Neoplasia 2019, 21, 571–581.
- Tanaka, Y.; Aoyagi, K.; Minashi, K.; Komatsuzaki, R.; Komatsu, M.; Chiwaki, F.; Tamaoki, M.; Nishimura, T.; Takahashi, N.; Oda, I.; et al. Discovery of a Good Responder Subtype of Esophageal Squamous Cell Carcinoma with Cytotoxic T-Lymphocyte Signatures Activated by Chemoradiotherapy. PLoS ONE 2015, 10, e0143804.
- Malhotra, G.K.; Yanala, U.; Ravipati, A.; Follet, M.; Vijayakumar, M.; Are, C. Global trends in esophageal cancer. J. Surg. Oncol. 2017, 115, 564–579.
- Wheeler, J.B.; Reed, C.E. Epidemiology of esophageal cancer. Surg. Clin. N. Am. 2012, 92, 1077–1087.
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28.
- Zheng, X.; Zhang, G.; Li, P.; Zhang, M.; Yan, X.; Zhang, X.; Yang, J.; Li, H.; Liu, X.; Ma, Z.; et al. Mutation tracking of a patient with EGFR-mutant lung cancer harboring de novo MET amplification: Successful treatment with gefitinib and crizotinib. Lung Cancer 2019, 129, 72–74.
- Merlo, L.M.; Pepper, J.W.; Reid, B.J.; Maley, C.C. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 2006, 6, 924–935.
- Michor, F.; Iwasa, Y.; Nowak, M.A. Dynamics of cancer progression. Nat. Rev. Cancer 2004, 4, 197–205.
- Bedard, P.L.; Hansen, A.R.; Ratain, M.J.; Siu, L.L. Tumour heterogeneity in the clinic. Nature 2013, 501, 355–364.
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta 2010, 1805, 105–117.
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94.
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavare, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014.
- Zhang, J.; Fujimoto, J.; Zhang, J.; Wedge, D.C.; Song, X.; Zhang, J.; Seth, S.; Chow, C.W.; Cao, Y.; Gumbs, C.; et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014, 346, 256–259.
- Xue, R.; Li, R.; Guo, H.; Guo, L.; Su, Z.; Ni, X.; Qi, L.; Zhang, T.; Li, Q.; Zhang, Z.; et al. Variable Intra-Tumor Genomic Heterogeneity of Multiple Lesions in Patients With Hepatocellular Carcinoma. Gastroenterology 2016, 150, 998–1008.
- Gerlinger, M.; Horswell, S.; Larkin, J.; Rowan, A.J.; Salm, M.P.; Varela, I.; Fisher, R.; McGranahan, N.; Matthews, N.; Santos, C.R.; et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat. Genet. 2014, 46, 225–233.
- Cao, W.; Wu, W.; Yan, M.; Tian, F.; Ma, C.; Zhang, Q.; Li, X.; Han, P.; Liu, Z.; Gu, J.; et al. Multiple region whole-exome sequencing reveals dramatically evolving intratumor genomic heterogeneity in esophageal squamous cell carcinoma. Oncogenesis 2015, 4, e175.
- Hao, J.J.; Lin, D.C.; Dinh, H.Q.; Mayakonda, A.; Jiang, Y.Y.; Chang, C.; Jiang, Y.; Lu, C.C.; Shi, Z.Z.; Xu, X.; et al. Spatial intratumoral heterogeneity and temporal clonal evolution in esophageal squamous cell carcinoma. Nat. Genet. 2016, 48, 1500–1507.
- Chen, X.X.; Zhong, Q.; Liu, Y.; Yan, S.M.; Chen, Z.H.; Jin, S.Z.; Xia, T.L.; Li, R.Y.; Zhou, A.J.; Su, Z.; et al. Genomic comparison of esophageal squamous cell carcinoma and its precursor lesions by multi-region whole-exome sequencing. Nat. Commun. 2017, 8, 524.
- Yan, T.; Cui, H.; Zhou, Y.; Yang, B.; Kong, P.; Zhang, Y.; Liu, Y.; Wang, B.; Cheng, Y.; Li, J.; et al. Multi-region sequencing unveils novel actionable targets and spatial heterogeneity in esophageal squamous cell carcinoma. Nat. Commun. 2019, 10, 1670.
- Yokoyama, A.; Kakiuchi, N.; Yoshizato, T.; Nannya, Y.; Suzuki, H.; Takeuchi, Y.; Shiozawa, Y.; Sato, Y.; Aoki, K.; Kim, S.K.; et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 2019, 565, 312–317.
- Chen, Z.Y.; Zhong, W.Z.; Zhang, X.C.; Su, J.; Yang, X.N.; Chen, Z.H.; Yang, J.J.; Zhou, Q.; Yan, H.H.; An, S.J.; et al. EGFR mutation heterogeneity and the mixed response to EGFR tyrosine kinase inhibitors of lung adenocarcinomas. Oncologist 2012, 17, 978–985.
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957.
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043.
- Su, K.Y.; Chen, H.Y.; Li, K.C.; Kuo, M.L.; Yang, J.C.; Chan, W.K.; Ho, B.C.; Chang, G.C.; Shih, J.Y.; Yu, S.L.; et al. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J. Clin. Oncol. 2012, 30, 433–440.
- Hofmann, W.K.; Komor, M.; Wassmann, B.; Jones, L.C.; Gschaidmeier, H.; Hoelzer, D.; Koeffler, H.P.; Ottmann, O.G. Presence of the BCR-ABL mutation Glu255Lys prior to STI571 (imatinib) treatment in patients with Ph+ acute lymphoblastic leukemia. Blood 2003, 102, 659–661.
- Deininger, M. Resistance to imatinib: Mechanisms and management. J. Natl. Compr. Cancer Netw. 2005, 3, 757–768.
- Linev, A.J.; Ivanov, H.J.; Zhelyazkov, I.G.; Ivanova, H.; Goranova-Marinova, V.S.; Stoyanova, V.K. Mutations Associated with Imatinib Mesylate Resistance—Review. Folia Med. 2018, 60, 617–623.
- Pectasides, E. Immune checkpoint blockade in esophageal squamous cell carcinoma: Is it ready for prime time? J. Thorac. Dis. 2018, 10, 1276–1279.
- Kang, X.; Chen, K.; Li, Y.; Li, J.; D’Amico, T.A.; Chen, X. Personalized targeted therapy for esophageal squamous cell carcinoma. World J. Gastroenterol. 2015, 21, 7648–7658.
- Guagnano, V.; Kauffmann, A.; Wohrle, S.; Stamm, C.; Ito, M.; Barys, L.; Pornon, A.; Yao, Y.; Li, F.; Zhang, Y.; et al. FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discov. 2012, 2, 1118–1133.
- Ng, C.K.; Pemberton, H.N.; Reis-Filho, J.S. Breast cancer intratumor genetic heterogeneity: Causes and implications. Expert Rev. Anticancer Ther. 2012, 12, 1021–1032.
- Van Vlodrop, I.J.; Niessen, H.E.; Derks, S.; Baldewijns, M.M.; van Criekinge, W.; Herman, J.G.; van Engeland, M. Analysis of promoter CpG island hypermethylation in cancer: Location, location, location! Clin. Cancer Res. 2011, 17, 4225–4231.
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400.
- Farazi, T.A.; Hoell, J.I.; Morozov, P.; Tuschl, T. MicroRNAs in human cancer. Adv. Exp. Med. Biol. 2013, 774, 1–20.
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337.
- Podlaha, O.; Riester, M.; De, S.; Michor, F. Evolution of the cancer genome. Trends Genet. 2012, 28, 155–163.
- Tian, T.; Olson, S.; Whitacre, J.M.; Harding, A. The origins of cancer robustness and evolvability. Integr. Biol. 2011, 3, 17–30.
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33.
- Baylin, S.B.; Ohm, J.E. Epigenetic gene silencing in cancer—A mechanism for early oncogenic pathway addiction? Nat. Rev. Cancer 2006, 6, 107–116.
- Komatsu, M.; Sasaki, H. DNA methylation is a key factor in understanding differentiation phenotype in esophageal squamous cell carcinoma. Epigenomics 2014, 6, 567–569.
- Tamaoki, M.; Komatsuzaki, R.; Komatsu, M.; Minashi, K.; Aoyagi, K.; Nishimura, T.; Chiwaki, F.; Hiroki, T.; Daiko, H.; Morishita, K.; et al. Multiple roles of single-minded 2 in esophageal squamous cell carcinoma and its clinical implications. Cancer Sci. 2018, 109, 1121–1134.
- Nishimura, T.; Tamaoki, M.; Komatsuzaki, R.; Oue, N.; Taniguchi, H.; Komatsu, M.; Aoyagi, K.; Minashi, K.; Chiwaki, F.; Shinohara, H.; et al. SIX1 maintains tumor basal cells via transforming growth factor-beta pathway and associates with poor prognosis in esophageal cancer. Cancer Sci. 2017, 108, 216–225.
- Nakazato, H.; Takeshima, H.; Kishino, T.; Kubo, E.; Hattori, N.; Nakajima, T.; Yamashita, S.; Igaki, H.; Tachimori, Y.; Kuniyoshi, Y.; et al. Early-Stage Induction of SWI/SNF Mutations during Esophageal Squamous Cell Carcinogenesis. PLoS ONE 2016, 11, e0147372.
- Kashima, H.; Noma, K.; Ohara, T.; Kato, T.; Katsura, Y.; Komoto, S.; Sato, H.; Katsube, R.; Ninomiya, T.; Tazawa, H.; et al. Cancer-associated fibroblasts (CAFs) promote the lymph node metastasis of esophageal squamous cell carcinoma. Int. J. Cancer 2019, 144, 828–840.
- Zhang, C.; Huang, H.; Miao, Y.; Xiong, H.; Lu, Z. Clonal distribution and intratumour heterogeneity of the B-cell repertoire in oesophageal squamous cell carcinoma. J. Pathol. 2018, 246, 323–330.
- Xiao, J.; Yang, W.; Xu, B.; Zhu, H.; Zou, J.; Su, C.; Rong, J.; Wang, T.; Chen, Z. Expression of fibronectin in esophageal squamous cell carcinoma and its role in migration. BMC Cancer 2018, 18, 976.
- Zhang, H.; Yue, J.; Jiang, Z.; Zhou, R.; Xie, R.; Xu, Y.; Wu, S. CAF-secreted CXCL1 conferred radioresistance by regulating DNA damage response in a ROS-dependent manner in esophageal squamous cell carcinoma. Cell Death Dis. 2017, 8, e2790.
- Zhang, H.; Xie, C.; Yue, J.; Jiang, Z.; Zhou, R.; Xie, R.; Wang, Y.; Wu, S. Cancer-associated fibroblasts mediated chemoresistance by a FOXO1/TGFbeta1 signaling loop in esophageal squamous cell carcinoma. Mol. Carcinog. 2017, 56, 1150–1163.
- Xing, S.; Zheng, X.; Zeng, T.; Zeng, M.S.; Zhong, Q.; Cao, Y.S.; Pan, K.L.; Wei, C.; Hou, F.; Liu, W.L. Chitinase 3-like 1 secreted by peritumoral macrophages in esophageal squamous cell carcinoma is a favorable prognostic factor for survival. World J. Gastroenterol. 2017, 23, 7693–7704.
- Chen, Z.; Zhang, C.; Pan, Y.; Xu, R.; Xu, C.; Chen, Z.; Lu, Z.; Ke, Y. T cell receptor beta-chain repertoire analysis reveals intratumour heterogeneity of tumour-infiltrating lymphocytes in oesophageal squamous cell carcinoma. J. Pathol. 2016, 239, 450–458.
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354.
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322.
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313.
- Wu, X.; Northcott, P.A.; Dubuc, A.; Dupuy, A.J.; Shih, D.J.; Witt, H.; Croul, S.; Bouffet, E.; Fults, D.W.; Eberhart, C.G.; et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature 2012, 482, 529–533.
- Anderson, K.; Lutz, C.; van Delft, F.W.; Bateman, C.M.; Guo, Y.; Colman, S.M.; Kempski, H.; Moorman, A.V.; Titley, I.; Swansbury, J.; et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 2011, 469, 356–361.
- De Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014, 346, 251–256.
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101.
- Liu, X.; Zhang, M.; Ying, S.; Zhang, C.; Lin, R.; Zheng, J.; Zhang, G.; Tian, D.; Guo, Y.; Du, C.; et al. Genetic Alterations in Esophageal Tissues From Squamous Dysplasia to Carcinoma. Gastroenterology 2017, 153, 166–177.
More
Information
Subjects:
Oncology; Genetics & Heredity
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
3 times
(View History)
Update Date:
12 Oct 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No