Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Emiliano Fabiani | + 2121 word(s) | 2121 | 2021-08-26 09:37:31 | | | |
| 2 | Vivi Li | Meta information modification | 2121 | 2021-08-30 04:55:50 | | | | |
| 3 | Conner Chen | Meta information modification | 2121 | 2021-09-22 04:13:45 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Fabiani, E. Forkhead Box Proteins in AML. Encyclopedia. Available online: https://encyclopedia.pub/entry/13635 (accessed on 07 February 2026).
Fabiani E. Forkhead Box Proteins in AML. Encyclopedia. Available at: https://encyclopedia.pub/entry/13635. Accessed February 07, 2026.
Fabiani, Emiliano. "Forkhead Box Proteins in AML" Encyclopedia, https://encyclopedia.pub/entry/13635 (accessed February 07, 2026).
Fabiani, E. (2021, August 27). Forkhead Box Proteins in AML. In Encyclopedia. https://encyclopedia.pub/entry/13635
Fabiani, Emiliano. "Forkhead Box Proteins in AML." Encyclopedia. Web. 27 August, 2021.
Copy Citation
Forkhead box (FOX) proteins are a group of transcriptional factors implicated in different cellular functions such as differentiation, proliferation and senescence. A growing number of studies have focused on the relationship between FOX proteins and cancers, particularly hematological neoplasms such as acute myeloid leukemia (AML). FOX proteins are widely involved in AML biology, including leukemogenesis, relapse and drug sensitivity. Here we explore the role of FOX transcription factors in the major AML entities, according to “The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia”, and in the context of the most recurrent gene mutations identified in this heterogeneous disease.
forkhead box proteins
acute myeloid leukemia
chemoresistance
1. Introduction
Forkhead box (FOX) proteins are an extended group of transcriptional factors characterized by the presence of an evolutionary conserved DNA-binding domain (DBD) named “winged-helix” or “fork-head”. The family name “fork-head” derives by the first gene discovered in Drosophila Melanogaster (forkhead, fkh) by Weigel et al. [1] in 1989 and was inspired by the fork-headed appearance of the mutated insect embryos, whereas the “winged-helix” name of the characteristic DBD present in all family members was suggested by the butterfly-like appearance of its three-dimensional structure. The prototypical DBD consists of about one hundred amino acids that under physiological conditions give rise to three α-helices, three β-sheets and two ‘wing’ regions that flank the third β-sheet [2].
FOX family members are now categorized, on the basis of sequence homology, into nineteen subgroups, from FOXA to FOXS, and reach at today the number of at least 50 genes distributed almost on all human chromosomes, Figure 1 [3][4].
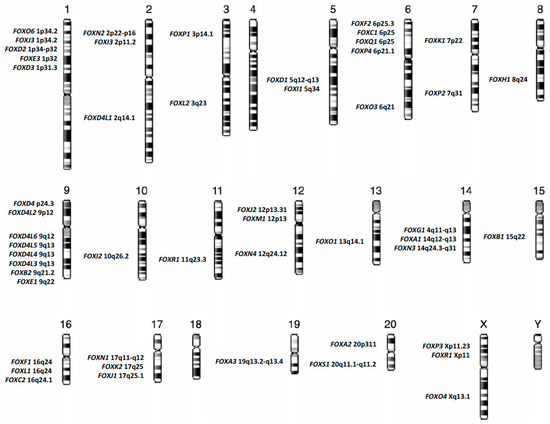
Figure 1. Chromosomal distribution of forkhead box (FOX) genes in the human genome.
Transcription factors are sequence-specific DNA-binding proteins (DBP) that control the rate of transcription of genetic information from DNA to messenger RNA, by binding to a specific DNA sequences (promoters and/or enhancers). As transcription factors, FOX proteins are responsible for the fine-tuning of gene expression during all stages of embryonic development and are guardians of the homeostasis in adult tissues. FOX proteins have been reported as active regulators of several networks, the main of which are: development, differentiation, maintenance of multipotency, proliferation, metabolism, DNA repair, cell cycle progression, migration, senescence, survival and apoptosis [5][6][7][8][9][10][11][12][13]. Despite the high sequence conservation of the forkhead domain, FOX proteins may exert different roles in the fine regulation of downstream genes, acting as repressors or activators of gene expression [14].
The mechanisms of gene expression regulation controlled by FOX proteins are, in some cases, so intricate that some FOX proteins are themselves the target of other members of the same gene family, as shown by Karadedou et al. that described the mechanisms by which FOXO3A and FOXM1 antagonize the activity of one another by regulating the transcription of downstream target genes [15]. The fine regulation of gene expression performed by FOX proteins is not only due to the tissue and/or cell-specific expression, but is also due to the post-translational modifications that mainly include phosphorylation, acetylation, ubiquitylation and sumoylation [16][17]. Post-translational modifications play a central role in cellular localization and activity of FOX factors. Mainly, FOX proteins act as transcriptional regulators in the nucleus, while they are prevalently inactive in the cytoplasm where they are subjected to proteasomal degradation.
The ability of FOX proteins to contribute to the control of several fundamental signaling pathways and of all the aspects of development and cell fate allows this superfamily of transcription factors to be heavily implicated in cancer initiation and progression. Indeed, FOX factors have been shown to play a role as either oncogenes or tumour suppressors, as well as active regulators of cellular resistance to chemotherapy and actionable targets in cancer therapy.
Myeloid neoplasms are a complex and heterogeneous group of hematopoietic diseases characterized by uncontrolled proliferation and/or blockage of differentiation of abnormal myeloid progenitor cells, and variable prognosis. “The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia” categorizes myeloid malignancies into five primary types: myeloproliferative neoplasms (MPN), myeloid/lymphoid neoplasms with eosinophilia and rearrangement of PDGFRA (platelet derived growth factor receptor alpha), PDGFRB (platelet derived growth factor receptor beta), or FGFR1 (fibroblast growth factor receptor 1), or with PCM1-JAK2 (pericentriolar material 1-Janus kinase 2), myelodysplastic/myeloproliferative neoplasms (MDS/MPN), myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) and related neoplasms [18].
Accumulating evidence suggests that FOX proteins are profoundly involved in the maintenance of multipotency of hematopoietic stem cells (HSC) and in critical mechanism driving aberrant self-renewal in preleukemic cells [19].
2. Forkhead Box Proteins in RUNX1-RUNX1T1 Acute Myeloid Leukemia
One of the most frequent initiating alterations in AML is the AML1-ETO translocation t(8;21), accounting for about 10% of total AML [20]. Although, according to “The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia” the t(8;21)(q22;q22.1), RUNX1-RUNX1T1 represents a specific subgroup of “AML with recurrent genetic abnormalities”, several authors have shown that the expression of RUNX1-RUNXT1 transcript in human hematopoietic stem progenitor cells (HSPC) causes deregulated differentiation and increased self-renewal of CD34+ cells without inducing AML [19][21]. Although, FOXO genes (FOXO1, FOXO3, FOXO4 and FOXO6) are frequently reported as tumour suppressors in several cancers [22][23][24][25][26][27][28][29][30], Lin et al. recently highlighted a new role of FOXO1 as an oncogene, clearly showing that the role of Forkhead box proteins depends on cellular context [19]. In this line, the Authors showed that the up-regulation of FOXO1 is required to sustain the growth of RUNX1-RUNXT1 cells, promoting the self-renewal and inhibiting the differentiation of human CD34+ HSPCs (Table 1). In particular, in RUNX1-RUNX1T1 CD34+ cells, increased levels of FOXO1 promote preleukemia transition and clonogenicity. Moreover, Lin et al. suggested the genetic and pharmacological ablation of FOXO1 as a therapeutic strategy for the elimination of preleukemic and leukemic t(8:21) HSPCs [19]. Indeed, an interesting therapeutic approach proposed for Acute Leukemia has been the interference with FOXO1 subcellular localization to improve blast cell sensitivity to antineoplastic drugs. Phosphorylation is the main cause of the 14-3-3 protein-mediated export of FOXO1 from an active nuclear form into the cytoplasm, where it results inactive [31][32]. Besides post-translational modifications, miRNA are emerging actors in regulating FOXOs levels and consequently AML blasts characteristics, such as drug sensitivity [33]. Together with FOXO3 activation, FOXO1 overexpression via miRNA interactions is involved in upregulation of ABCB1 gene, coding for P-glycoprotein, well known responsible for decreased drug accumulation in multidrug-resistant cancer cells [34].
Table 1. Forkhead box factors deregulation according to acute myeloid leukemia (AML) recurrent abnormalities and their involvement in biological processes.
| Recurrent Abnormalities in Acute Myeloid Leukemia | FOX Family Member | Biological Process | References |
|---|---|---|---|
| t(8;21); RUNX1-RUNX1T1 | FOXO1 | Self-renewal and Differentiation | Lin et al. [19] |
| PML-RARA | FOXO3A FOXC1 |
Apoptosis and Granulocytic Differentiation Granulocytic Differentiation and Epigenetic Regulation |
Sakoe et al. [35] Somerville et al. [36] and Fabiani et al. [37] |
| NPM1 | FOXM1 | Cell Proliferation, Division and Chemoresistance | Laoukili et al. [38] Nakamura et al. [39] Khan et al. [40] |
| FLT3 ITD | FOXO3A FOXO1 FOXM1 |
Apoptosis, Survival and Proliferation Cell growth, Apoptosis and antioxidant defences Survival, Apoptosis and Chemoresistance |
Scheijen et al. [41] Seedhouse et al. [42] Liu et al. [43] |
| IDH 1-2 | FOXOs | Cellular Differentiation and Tumor Suppression/Progression and Epigenetic Instability | Charitou et al. [44] |
3. Forkhead Box Proteins in PML/RARA (Promyelocytic Leukemia/Retinoic Acid Receptor-Alpha) Acute Promyelocytic Leukemia
Acute promyelocytic leukemia (APL) is another subgroup of acute myeloid leukemia characterized by a unique t(15;17) translocation generating the PML/RARA fusion gene.
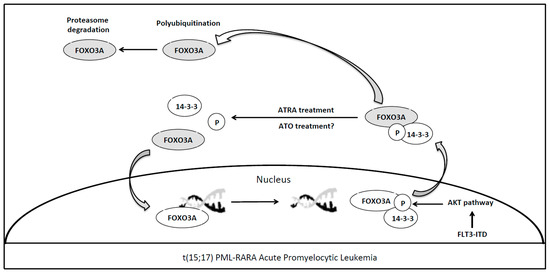
The PML/RARA oncoprotein synthesis is the key pathogenetic event of APL specifically targeted by all-trans retinoic acid (ATRA) and arsenic trioxide (ATO), two non-chemotherapeutic agents that synergistically act inducing oncoprotein degradation. Although it is well known that this fusion protein blocks granulocytic differentiation by direct transcriptional inhibition of retinoic acid target genes [45], the detailed mechanisms and the complete list of genes involved in APL transformation are not fully understood. Although, the ATRA-related apoptosis of APL blasts via tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression has been previously reported, the pivotal transcription factor has not been yet identified [46]. Sakoe et al. have shown that activation of FOXO3A is an essential event for ATRA-induced cellular response in human t(15:17) cell line NB4 [35]. Nuclear FOXO3A tunes HSC maintenance and, if phosphorylated, it is exported to the cytoplasm and interacts with 14-3-3 proteins, hence losing its function [47]. The phosphorylation of FOXO3A, and its consequent loss of function, is regulated by AKT pathway aberrantly activated in case of AML driver mutations such as FLT3-ITD or BCR-ABL [48], (Figure 2). In the APL setting, Sakoe et al. showed that ATRA treatment was able to reduce FOXO3A phosphorylation and to induce the translocation of the transcription factor in the nucleus, where the protein leads to apoptosis trough TRAIL upregulation [35]. Notably, FOXO3A silencing through shRNA inhibited ATRA-induced response in NB4 cells, as such as in ATRA resistant NB4/RA cells. ATRA treatment was unable to induce FOXO3A phosphorylation, TRAIL upregulation, apoptosis and granulocytic differentiation; whereas forced expression of active FOXO3A in the nucleus induced TRAIL production and apoptosis in NB4/RA cells [35] (Table 1). Thus, in APL, a member of FOXOs family acts as tumor suppressor gene differently from the previously reported role of FOXO1 in RUNX1-RUNX1T1 acute myeloid leukemia. These results highlight a possible role for FOXO3A as a potential therapeutic target in APL to overcome ATRA resistance.
Figure 2. ATRA mediated reactivation of FOXO3A in t(15;17) PML-RARA acute promyelocytic leukemia. Nuclear FOXO3A (active form), once phosphorylated, interacting with 14-3-3 protein is exported to the cytoplasm losing its function [47]. The phosphorylation of FOXO3A, and its consequent loss of function, may be regulated by AKT pathway aberrantly activated in case of AML driver mutations such as FLT3-ITD [48]. All-trans retinoic acid (ATRA) treatment is able to reduce FOXO3A phosphorylation and to induce the relocation of the transcription factor into the nucleus, where the protein leads to blast apoptosis [35]. Arsenic trioxide (ATO) treatment in this cellular context needs to be better assessed. Light grey shows the FOXO3A inactive form.
Arsenic trioxide in combination with ATRA is currently considered the standard of care for adults with low-to-intermediate-risk APL, with a complete remission rate near to 100% [49]. Interestingly, Zhang et al. have recently shown that ATO treatment of gastric cancer cells induced the upregulation of FOXO3A expression in the nucleus and that FOXO3A knockdown attenuated the effect of ATO treatment in gastric cancer cells and in mouse models [50]. Moreover, they also demonstrate that ATO-related nuclear upregulation of active FOXO3A is the result of its phosphorylation via the aforementioned AKT pathway, leading to inhibition of cell migration. This new insight may be useful for further investigations about the role of FOXO3A in treatment response to ATO in APL patients. In this line, additional studies to identify novel therapeutic agents enabled to restore FOXO3A function may overcome ATO resistance even in patients harboring PML-A216V mutation, accounting for about 30% of ATO-resistant cases [51].
FOXO subfamily members are not the solely forkhead transcription factors involved in APL pathogenesis. In 2015, Somerville et al. reported the overexpression of FOXC1 in nearly 20% of primary non-APL AML samples, showing its involvement in the monocyte/macrophage differentiation block and in the increased clonogenic potential of AML cells [36]. FOXC1 is deregulated in different types of cancers and is often associated with poor prognosis in AML, cooperating with HOXA/B and consequently repressing the monocyte transcriptional regulator KLF4 [36]. More recently, our group showed that FOXC1 mRNA and protein levels were significantly lower in primary marrow samples from APL patients, as compared to samples obtained from patients with other AML subtypes, and normal CD34+ hematopoietic cells [37]. Moreover, we demonstrated that FOXC1 expression was significantly increased in APL samples following consolidation treatment and that ATRA treatment unlocked FOXC1 expression in NB4, but not in NB4-R4 ATRA-resistant cells. Of note, using chromatin immune precipitation assay (ChIP), we identified functional binding sites of ATRA in the FOXC1 promoter region. In addition, we showed that in diagnostic APL samples and in NB4 cells, reduced FOXC1 expression was associated to DNA hypermethylation of the +354 to +568 FOXC1 region and that hypomethylating treatment with decitabine of NB4 upregulated FOXC1 expression [37] (Table 1). Our findings indicate a dual repression model of FOXC1 expression in APL giving the rationale for a potential role of hypomethylating treatment (HMT) in advanced and/or resistant APL. HMT is anecdotally reported for the treatment of resistant APL cases and the rationale for their use has been postulated by the study of Soncini et al. where they show TRAIL-dependent apoptosis of APL and AML blasts [52]. In this work Soncini et al. identify a TRAIL promoter region, affected by DNA hypermethylation and whose function is restored after decitabine administration, demonstrating in APL a novel therapeutic approach other than PML-RARA degradation (ATRA-ATO scheme), using epigenetic drugs [52]. This strategy may be helpful to overcome resistance to differentiating treatment in case of mutations in PML or RARA.
References
- Weigel, D.; Jürgens, G.; Küttner, F.; Seifert, E.; Jäckle, H. The homeotic gene fork head encodes a nuclear protein and is expressed in the terminal regions of the Drosophila embryo. Cell 1989, 57, 645–658.
- Hannenhalli, S.; Kaestner, K.H. The evolution of Fox genes and their role in development and disease. Nat. Rev. Genet. 2009, 10, 233–240.
- Jackson, B.C.; Carpenter, C.; Nebert, D.W.; Vasiliou, V. Update of human and mouse forkhead box (FOX) gene families. Hum. Genom. 2010, 4, 345–352.
- Wang, J.; Li, W.; Zhao, K.; Fu, W.; Zheng, X.; Pang, X.; Du, G. Members of FOX family could be drug targets of cancers. Pharmacol. Ther. 2018, 181, 183–196.
- Golson, M.L.; Kaestner, K.H. Fox transcription factors: From development to disease. Development 2016, 143, 4558–4570.
- He, L.; Gomes, A.P.; Wang, X.; Yoon, S.O.; Lee, G.; Nagiec, M.J.; Cho, S.; Chavez, A.; Islam, T.; Yu, Y.; et al. mTORC1 Promotes Metabolic Reprogramming by the Suppression of GSK3-Dependent Foxk1 Phosphorylation. Mol. Cell 2018, 70, 949–960.
- Maachani, U.B.; Shankavaram, U.; Kramp, T.; Tofilon, P.; Camphausen, K.; Tandle, A.T. FOXM1 and STAT3 interaction confers radioresistance in glioblastoma cells. Oncotarget 2016, 7, 77365–77377.
- Alvarez-Fernández, M.; Medema, R.H. Novel functions of FoxM1: From molecular mechanisms to cancer therapy. Front. Oncol. 2013, 3, 30.
- Ackermann, S.; Kocak, H.; Hero, B.; Ehemann, V.; Kahlert, Y.; Oberthuer, A.; Roels, F.; Theißen, J.; Odenthal, M.; Berthold, F.; et al. FOXP1 inhibits cell growth and attenuates tumorigenicity of neuroblastoma. BMC Cancer 2014, 14, 840.
- Wang, P.; Lv, C.; Zhang, T.; Liu, J.; Yang, J.; Guan, F.; Hong, T. FOXQ1 regulates senescence-associated inflammation via activation of SIRT1 expression. Cell Death Dis. 2017, 8, e2946.
- Macedo, J.C.; Vaz, S.; Bakker, B.; Ribeiro, R.; Bakker, P.L.; Escandell, J.M.; Ferreira, M.G.; Medema, R.; Foijer, F.; Logarinho, E. FoxM1 repression during human aging leads to mitotic decline and aneuploidy-driven full senescence. Nat. Commun. 2018, 9, 2834.
- Song, B.N.; Chu, I.S. A gene expression signature of FOXM1 predicts the prognosis of hepatocellular carcinoma. Exp. Mol. Med. 2018, 50, e418.
- Gao, Y.F.; Zhu, T.; Mao, X.Y.; Mao, C.X.; Li, L.; Yin, J.Y.; Zhou, H.H.; Liu, Z.Q. Silencing of Forkhead box D1 inhibits proliferation and migration in glioma cells. Oncol. Rep. 2017, 37, 1196–1202.
- Lam, E.W.; Brosens, J.J.; Gomes, A.R.; Koo, C.Y. Forkhead box proteins: Tuning forks for transcriptional harmony. Nat. Rev. Cancer 2013, 13, 482–495.
- Karadedou, C.T.; Gomes, A.R.; Chen, J.; Petkovic, M.; Ho, K.K.; Zwolinska, A.K.; Feltes, A.; Wong, S.Y.; Chan, K.Y.; Cheung, Y.N.; et al. FOXO3a represses VEGF expression through FOXM1-dependent and -independent mechanisms in breast cancer. Oncogene 2012, 31, 1845–1858.
- Rocca, D.L.; Wilkinson, K.A.; Henley, J.M. SUMOylation of FOXP1 regulates transcriptional repression via CtBP1 to drive dendritic morphogenesis. Sci. Rep. 2017, 7, 877.
- Myatt, S.S.; Lam, E.W. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 2007, 7, 847–859.
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405.
- Lin, S.; Ptasinska, A.; Chen, X.; Shrestha, M.; Assi, S.A.; Chin, P.S.; Imperato, M.R.; Aronow, B.J.; Zhang, J.; Weirauch, M.T.; et al. A FOXO1-induced oncogenic network defines the AML1-ETO preleukemic program. Blood 2017, 130, 1213–1222.
- Müller, A.M.; Duque, J.; Shizuru, J.A.; Lübbert, M. Complementing mutations in core binding factor leukemias: From mouse models to clinical applications. Oncogene 2008, 27, 5759–5773.
- Tonks, A.; Pearn, L.; Musson, M.; Gilkes, A.; Mills, K.I.; Burnett, A.K.; Darley, R.L. Transcriptional dysregulation mediated by RUNX1-RUNX1T1 in normal human progenitor cells and in acute myeloid leukaemia. Leukemia 2007, 21, 2495–2505.
- Jagani, Z.; Singh, A.; Khosravi-Far, R. FoxO tumor suppressors and BCR-ABL-induced leukemia: A matter of evasion of apoptosis. Biochim. Biophys. Acta 2008, 1785, 63–84.
- Kornblau, S.M.; Singh, N.; Qiu, Y.; Chen, W.; Zhang, N.; Coombes, K.R. Highly phosphorylated FOXO3A is an adverse prognostic factor in acute myeloid leukemia. Clin. Cancer Res. 2010, 16, 1865–1874.
- Obrador-Hevia, A.; Serra-Sitjar, M.; Rodríguez, J.; Villalonga, P.; Fernández de Mattos, S. The tumour suppressor FOXO3 is a key regulator of mantle cell lymphoma proliferation and survival. Br. J. Haematol. 2012, 156, 334–345.
- Xie, L.; Ushmorov, A.; Leithäuser, F.; Guan, H.; Steidl, C.; Färbinger, J.; Pelzer, C.; Vogel, M.J.; Maier, H.J.; Gascoyne, R.D.; et al. FOXO1 is a tumor suppressor in classical Hodgkin lymphoma. Blood 2012, 119, 3503–3511.
- Yao, S.; Mahmud, Z.; Sachini, N.; Aimjongjun, S.; Saavedra-García, P.; Lam, E.W. Characterization of FOXO Acetylation. Methods Mol. Biol. 2019, 1890, 77–90.
- Rehman, A.; Kim, Y.; Kim, H.; Sim, J.; Ahn, H.; Chung, M.S.; Shin, S.J.; Jang, K. FOXO3a expression is associated with lymph node metastasis and poor disease-free survival in triple-negative breast cancer. J. Clin. Pathol. 2018, 71, 806–813.
- Hou, T.; Li, Z.; Zhao, Y.; Zhu, W.G. Mechanisms controlling the anti-neoplastic functions of FoxO proteins. Semin. Cancer Biol. 2018, 50, 101–114.
- Pan, C.W.; Jin, X.; Zhao, Y.; Pan, Y.; Yang, J.; Karnes, R.J.; Zhang, J.; Wang, L.; Huang, H. AKT-phosphorylated FOXO1 suppresses ERK activation and chemoresistance by disrupting IQGAP1-MAPK interaction. EMBO J. 2017, 36, 995–1010.
- Zhu, H. Targeting forkhead box transcription factors FOXM1 and FOXO in leukemia. Oncol. Rep. 2014, 32, 1327–1334.
- Farhan, M.; Wang, H.; Gaur, U.; Little, P.J.; Xu, J.; Zheng, W. FOXO Signaling Pathways as Therapeutic Targets in Cancer. Int. J. Biol. Sci. 2017, 13, 815–827.
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288.
- Hui, R.C.; Francis, R.E.; Guest, S.K.; Costa, J.R.; Gomes, A.R.; Myatt, S.S.; Brosens, J.J.; Lam, E.W. Doxorubicin activates FOXO3a to induce the expression of multidrug resistance gene ABCB1 (MDR1) in K562 leukemic cells. Mol. Cancer Ther. 2008, 7, 670–678.
- Han, C.Y.; Cho, K.B.; Choi, H.S.; Han, H.K.; Kang, K.W. Role of FoxO1 activation in MDR1 expression in adriamycin-resistant breast cancer cells. Carcinogenesis 2008, 29, 1837–1844.
- Sakoe, Y.; Sakoe, K.; Kirito, K.; Ozawa, K.; Komatsu, N. FOXO3A as a key molecule for all-trans retinoic acid–induced granulocytic differentiation and apoptosis in acute promyelocytic leukemia. Blood 2010, 115, 3787–3795.
- Somerville, T.D.; Wiseman, D.H.; Spencer, G.J.; Huang, X.; Lynch, J.T.; Leong, H.S.; Williams, E.L.; Cheesman, E.; Somervaille, T.C. Frequent Derepression of the Mesenchymal Transcription Factor Gene FOXC1 in Acute Myeloid Leukemia. Cancer Cell 2015, 28, 329–342.
- Fabiani, E.; Falconi, G.; Noguera, N.I.; Saulle, E.; Cicconi, L.; Divona, M.; Banella, C.; Picardi, A.; Cerio, AM.; Boe, L.; et al. The forkhead box C1 (FOXC1) transcription factor is downregulated in acute promyelocytic leukemia. Oncotarget 2017, 8, 84074–84085.
- Laoukili, J.; Stahl, M.; Medema, R.H. FoxM1: At the crossroads of ageing and cancer. Biochim. Biophys. Acta 2007, 1775, 92–102.
- Nakamura, S.; Hirano, I.; Okinaka, K.; Takemura, T.; Yokota, D.; Ono, T.; Shigeno, K.; Shibata, K.; Fujisawa, S.; Ohnishi, K. The FOXM1 transcriptional factor promotes the proliferation of leukemia cells through modulation of cell cycle progression in acute myeloid leukemia. Carcinogenesis 2010, 31, 2012–2021.
- Khan, I.; Halasi, M.; Patel, A.; Schultz, R.; Kalakota, N.; Chen, Y.H.; Aardsma, N.; Liu, L.; Crispino, J.D.; Mahmud, N.; et al. FOXM1 contributes to treatment failure in acute myeloid leukemia. JCI Insight 2018, 3.
- Scheijen, B.; Ngo, H.T.; Kang, H.; Griffin, J.D. FLT3 receptors with internal tandem duplications promote cell viability and proliferation by signaling through Foxo proteins. Oncogene 2004, 23, 3338–3349.
- Seedhouse, C.H.; Mills, K.I.; Ahluwalia, S.; Grundy, M.; Shang, S.; Burnett, A.K.; Russell, N.H.; Pallis, M. Distinct poor prognostic subgroups of acute myeloid leukaemia, FLT3-ITD and P-glycoprotein-positive, have contrasting levels of FOXO1. Leuk. Res. 2014, 38, 131–137.
- Liu, L.L.; Zhang, D.H.; Mao, X.; Zhang, X.H.; Zhang, B. Over-expression of FoxM1 is associated with adverse prognosis and FLT3-ITD in acute myeloid leukemia. Biochem. Biophys. Res. Commun. 2014, 446, 280–285.
- Charitou, P.; Rodriguez-Colman, M.; Gerrits, J.; van Triest, M.; Groot Koerkamp, M.; Hornsveld, M.; Holstege, F.; Verhoeven-Duif, N.M.; Burgering, B.M. FOXOs support the metabolic requirements of normal and tumor cells by promoting IDH1 expression. EMBO Rep. 2015, 16, 456–466.
- Grignani, F.; Ferrucci, P.F.; Testa, U.; Talamo, G.; Fagioli, M.; Alcalay, M.; Mencarelli, A.; Grignani, F.; Peschle, C.; Nicoletti, I.; et al. The acute promyelocytic leukemia-specific PML-RAR alpha fusion protein inhibits differentiation and promotes survival of myeloid precursor cells. Cell 1993, 74, 423–431.
- Altucci, L.; Rossin, A.; Raffelsberger, W.; Reitmair, A.; Chomienne, C.; Gronemeyer, H. Retinoic acid-induced apoptosis in leukemia cells is mediated by paracrine action of tumor-selective death ligand TRAIL. Nat. Med. 2001, 7, 680–686.
- Birkenkamp, K.U.; Coffer, P.J. Regulation of cell survival and proliferation by the FOXO (Forkhead box, class O) subfamily of Forkhead transcription factors. Biochem. Soc. Trans. 2003, 31, 292–297.
- Komatsu, N.; Watanabe, T.; Uchida, M.; Mori, M.; Kirito, K.; Kikuchi, S.; Liu, Q.; Tauchi, T.; Miyazawa, K.; Endo, H.; et al. A member of Forkhead transcription factor FKHRL1 is a downstream effector of STI571-induced cell cycle arrest in BCR-ABL-expressing cells. J. Biol. Chem. 2003, 278, 6411–6419.
- Lo-Coco, F.; Avvisati, G.; Vignetti, M.; Thiede, C.; Orlando, S.M.; Iacobelli, S.; Ferrara, F.; Fazi, P.; Cicconi, L.; Di Bona, E.; et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N. Engl. J. Med. 2013, 369, 111–121.
- Zhang, L.; Liu, L.; Zhan, S.; Chen, L.; Wang, Y.; Zhang, Y.; Du, J.; Wu, Y.; Gu, L. Arsenic Trioxide Suppressed Migration and Angiogenesis by Targeting FOXO3a in Gastric Cancer Cells. Int. J. Mol. Sci. 2018, 19.
- Alfonso, V.; Iaccarino, L.; Ottone, T.; Cicconi, L.; Lavorgna, S.; Divona, M.; Cairoli, R.; Cristiano, A.; Ciardi, C.; Travaglini, S.; et al. Early and sensitive detection of PML-A216V mutation by droplet digital PCR in ATO-resistant acute promyelocytic leukemia. Leukemia 2019.
- Soncini, M.; Santoro, F.; Gutierrez, A.; Frigè, G.; Romanenghi, M.; Botrugno, O.A.; Pallavicini, I.; Pelicci, P.; Di Croce, L.; Minucci, S. The DNA demethylating agent decitabine activates the TRAIL pathway and induces apoptosis in acute myeloid leukemia. Biochim. Biophys. Acta 2013, 1832, 114–120.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
757
Revisions:
3 times
(View History)
Update Date:
22 Sep 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No