Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Manuel Glauco Carbone | + 3040 word(s) | 3040 | 2021-08-24 07:41:45 | | | |
| 2 | Lindsay Dong | Meta information modification | 3040 | 2021-08-26 03:16:32 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Carbone, M.G. Platelet APP in Alzheimer’s Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/13518 (accessed on 07 February 2026).
Carbone MG. Platelet APP in Alzheimer’s Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/13518. Accessed February 07, 2026.
Carbone, Manuel Glauco. "Platelet APP in Alzheimer’s Disease" Encyclopedia, https://encyclopedia.pub/entry/13518 (accessed February 07, 2026).
Carbone, M.G. (2021, August 24). Platelet APP in Alzheimer’s Disease. In Encyclopedia. https://encyclopedia.pub/entry/13518
Carbone, Manuel Glauco. "Platelet APP in Alzheimer’s Disease." Encyclopedia. Web. 24 August, 2021.
Copy Citation
Alzheimer’s disease (AD) is a multifactorial age-related progressive neurodegenerative disorder characterized by gradual memory loss, cognitive decline and functional alteration that cause difficulties in the performance of everyday life activities and loss of self-identity.
Alzheimer’s disease
Aβ cascade
platelet activation
APP processing
Aβ amyloid
1. Introduction
1.1. Alzheimer’s Disease
The most evident macroscopic characteristic of the brain of a subject suffering from Alzheimer’s disease is the marked cortical atrophy that determining increased amplitude of the cerebral furrows and the increase in the ventricular volume. This atrophy is diffuse, affecting, in addition to the temporal lobe, the cortical associative areas, the hippocampus and the para-hippocampal gyrus, with a relative saving of the posterior areas of the hemispheres, of the cerebellum and of the brain stem. Atrophy is mainly linked to neuronal degeneration, which involves a reduction in the number of dendritic spines and synaptic junctions. Among the subcortical structures, particularly affected are the amygdala, the locus coeruleus, the raphe nucleus and the cholinergic structures of the brain stem, these alterations correlate with the course and extent of the disease [1]. Alzheimer’s disease is thought to begin 20 years or more before symptoms arise, with small changes in the brain that are unnoticeable to the person affected [2][3][4][5][6][7][8].
1.2. The Diagnosis Limits
Currently, AD pathological diagnosis is based on the pathology of the post-mortem, which is marked with extracellular age pigment, intracellular nerve fiber tangles in the hippocampal and/or cortical regions, as well as a significant reduction in the gray matter [9]. Because of the pervasiveness of AD pathology in the elderly, biomarkers have become an essential component of Alzheimer disease (AD) research and a potential tool for the diagnosis of AD at the preclinical stage. It was proposed the “A/T/N” system in which seven major AD biomarkers are divided into three binary categories based on the nature of the pathophysiology that each measure. “A” refers to the value of a β-amyloid biomarker (amyloid PET or CSF Aβ42); “T” the value of a tau biomarker (CSF p-tau, or tau PET); and “N” biomarkers of neurodegeneration or neuronal injury ([18F]-fluorodeoxyglucose–PET, structural MRI, or CSF total tau) [10]. Positron-emission tomography (PET) imaging has been applied to the detection of Aβ in the brain and has revealed that Aβ peptide accumulates in the frontal cortex of patients with mild cognitive impairment (MCI), the prodromal stage of AD [11][12][13]. Thus, PET imaging of Aβ represents a promising tool for the early diagnosis of AD, but it is a sophisticated technique that requires special equipment and cannot be widely used. Low CSF Aβ42 levels reflect the decreased clearance of Aβ42 and its deposition in the brain, but this is not absolutely specific for AD and is also observed in patients with dementia with Lewy bodies. Elevated phosphorylated tau (p-tau) is a more specific marker, and measurements of either p181-tau, or p231-tau give similar diagnosis accuracy [14][15]. The combination of Aβ42, total tau and p-tau provides a diagnosis for AD with a sensitivity of 80% and a specificity of 90% and can help predict the conversion from MCI to AD [16][17]. However, these markers remain insufficiently used due to the delicate procedure of CSF collection by lumbar puncture. As compared to CSF-based biomarkers, which undoubtedly bear a closer relationship with the abnormalities that occur in the brain [18], the search for peripheral biomarkers of AD is justified by its better accessibility and tolerability, i.e., samples can be obtained by less invasive procedures [19]. In addition, blood-based biomarkers may be more adequate for longitudinal studies that require multiple sampling [20], but most of the available data present inconsistency and lack absolute specificity and sensitivity [21][22]. Platelets are considered the most accessible peripheral neuronal-like cellular system and have been suggested as a promising model since they are the major peripheral reserve of amyloid precursor protein (APP) providing over 90% of blood Aβ [23][24][25].
1.3. The APP Processing Phase
APP is an integral Type-I transmembrane protein present in several cell types [26][27][28][29][30][31]. It is concentrated in synapses and takes part in cell-matrix and cell-cell interaction in neurons [32][33]. This adhesion molecule also participates in various processes in different tissues, for example, APP is involved in hemostasis, thrombosis, sperm motility and sperm-oocyte interaction [34][35]. The Aβ hypothesis was formulated, suggesting that an imbalance between production and clearance of Aβ (Aβ dyshomeostasis) is an early, often initiating factor in AD [36]. However, Aβ plaques were sometimes present in cognitively normal individuals and in the meanwhile neuronal death also occurred in brain regions devoid of plaques [37]. Oligomers of Aβ peptides are toxic to brain cells and there is no direct correlation between the manifestation of the disease and plaque burden [38]. The most common view is that increased concentrations of Aβ oligomers trigger neuronal dysfunction and network alterations, with secondary damage produced by hyperphosphorylated tau protein aggregated in tangles [39][40]. APP exists in several alternatively spliced isoforms, APP695, APP751, and APP770. The major APP isoforms result from alternative splicing of exon 7 that encodes a Kunitz serine protease inhibitor domain (KPI), exon 8 that codes for a domain with homology to the MRC OX-2 antigen (OX-2) and exon 15. The APP695 isoform, which lacks the KPI (APP-KPI) and OX-2 domains, is expressed predominantly in neuronal cells. Peripheral cells and platelets, preferably express APP isoforms that contain the KPI domain (APP-KPI+), including APP751 (lacking the OX-2 domain) and APP770 (expressing all exons) [41][42][43][44]. APP is cleaved by sequential actions of α-, β-, and γ-secretases [45]. Most of the APP protein is processed by α-secretases in the non-amyloidogenic pathway, which involves cleavage within the Aβ sequence [46]. α-secretase enzymes belong to the family of disintegrin and metalloprotease including ADAM-10 and ADAM-17 [47][48].This process takes place in the secretory pathway, at the plasma membrane and in secretory vesicles. ADAM-10 exerts the major part of the α-secretase activity. It generates the neuroprotective and neurotrophic soluble ectodomain fragment 100–130 kDa (sAPP-α) and non-neurotoxic membrane-associated carboxy-terminal fragments (CTFα or C83) [49][50][51][52][53]. Alternatively, APP is processed by β-secretase at the amino terminus of Aβ parts releasing the soluble N-terminal fragment, sAPP-β and a carboxy-terminal fragment (CTFβ or C-99) through the amyloidogenic pathway [54][55]. β-site APP-cleaving enzyme 1 (BACE1) is a Type I transmembrane aspartic proteases and has been reported to exert β-secretase activity [56]. APP CTFα/β is cleaved at the ε-site by the γ-secretase complex, a membrane-embedded multimeric aspartic protease comprising presenilin 1 or 2, nicastrin (NCT), anterior pharynx defective 1 (APH-1), and presenilin enhancer 2 [57].The γ-secretase action bring to the release of the carboxy-terminal half of APP CTFs, APP intracellular domain (AICD), into the cytosol (6, 7) and secretes the amino-terminal half of APP CTFα/β, p3 and Aβ from APP CTFα and CTFβ respectively [58][59][60][61]. Following the primary ε-cleavage, further cleavage of the amino-terminal half of APP CTFα/β at multiple γ-sites occurs, and various neurotoxic species of Aβ including Aβ49, Aβ46, Aβ43, and ultimately Aβ40, the major Aβ species, are generated from APP CTFβ [62]. Alternative cleavage of CTFβ at the minor ε-site results in Aβ48, Aβ45, Aβ42, and, finally, Aβ38, which does not aggregate and is not neurotoxic [63][64]. In contrast to neurons that predominantly process APP via the β-secretase pathway, platelets, like other non-neuronal cells, process APP mostly through α-secretase. It has been shown that sAPP concentrations in platelets are much higher than Aβ peptides [65].
2. Platelet APP Processing in Alzheimer’s Disease
2.1. Amyloid Protein Precursor (APP)
Delineation of the mechanisms involved in APP trafficking is thus relevant and crucial to understanding the pathogenesis of AD. One of the first milestones in the comprehension of the pathogenesis of AD dates to 1984 with the works of Glenner and Masters, when cerebral Aβ deposits in senile and neuritic plaques were recognized as playing a central role [66][67][68][69]. Afterwards, from the first study conducted by Bush et al. in 1990, questions are raised about the strength of platelet APP as a peripheral biomarker of AD and as a potential therapeutic target [70]. Bush was the first to show that APP is released by platelets and, although failing to find any differences on the APP isoforms expression between AD and controls, pointed out the possibility of a relationship between APP processing and AD. They hypothesized a possible vescicular release of platelet APP that raises the probability of circulating form of APP being the substrate for the proteolytic events that result in the production of Aβ [70]. Specifically, they found a 50% increase in the proportion of 130 kDa APP species in AD and a 20–35% decrease in the proportion of 42 kDa APP. The comparison of the 130 kDa plasma APP levels in AD patients (moderate and severe grade) with those of control subjects allowed to distinguish these groups with a specificity of 87.0% and a sensitivity of 79.4% [71].
Contrary, Davies et al. (1993 and 1997) showed that AD patients’ platelets activated by α-thrombin, compared to those of controls and to those of patients with other brain neurodegenerative diseases (the groups were not matched by age and gender), tended to abnormally hyperacidify, to accumulate unprocessed 120–130 kDa APP on their surface and to release less sAPP. These changes were observed only in patients with advanced AD suggesting that the hypothetical platelet defect appeared in the late stages of disease [72][73][74].
In line with these findings, APP ratio (APPr = APP130/APP106 − 110) was found to be significantly lower in patients with AD compared to age-matched controls and to individuals with neurocognitive disorders not AD related [75]. Furthermore, APPr in patients with AD significantly correlated with the progression and the severity of the disease [76][77][78]. Unlike Davies et al., the differences in the APP processing are evident in subjects with mild AD [76]. They also found no difference in APP mRNA transcripts levels between experimental groups, a fact that may suggest the abnormal proteolytic processing of platelet APP in AD [76]. These findings were further replicated and were independently from age and ApoE4 carrier status [31][77][79][80][81][82][83]. For the first time, it was hypothesized a platelet hyperactivation state or a platelet hyper-responsivity as the main cause for APP processing alterations and therefore for abnormal Aβ production [79][80].
It was also associated to the APP decrease a significant reduction of the platelet ADAM-10 activity, parallel to reduced plasma and CSF α-APPs, or increased levels of Aβ and a heightened activity of the active BACE-1 forms [84][85][86]. The preclinical diagnostic value of APPr could be even enhanced when combined with measurement of regional cerebral blood flow by SPECT scan. The positive predictive value of these combined markers in identifying progressive MCI was 0.87, and the negative predictive value was 0.90 [87]. Furthermore, to improve the diagnostic specificity with the key-element of beta-amyloid cascade it was used an artificial neural networks (ANNs) to afford non-linear tasks, and with the best ANN model they correctly identified mild AD patients in the 94% of cases and control subjects in the 92% [88].
Attempting to find a reliable peripheral biomarker for the diagnosis of AD, Vignini et al. (2013) examined the platelet APP isoform mRNAs using the real-time quantitative PCR. The gene expression measurements in the AD patient group revealed a significant up-regulation of APP TOT (1.52-fold), APP KPI (1.32-fold), APP 770 (1.33-fold) and APP 751 (1.26-fold) compared to controls. Moreover, a statistically significant positive correlation was found between APP mRNA levels (TOT, KPI, 770 and 751) and cognitive impairment [89]. These findings were replicated in another study in which AD patients were compared to front-temporal lobar dementia (FTLD) and controls. They found a significant up-regulation of APP TOT and APP KPI in both AD and FTLD patients compared to the controls, although the severity of cognitive decline did not correlate with the expression of up-regulation in FTLD patients [90].
Finally, one study did not any find any differences in APP isoform expressions between AD patients and control groups [91].
2.2. The APP Processing System
The α- and β-secretase activity has so far been investigated using different methodologies and has been correlated to the APPr and the degree of cognitive impairment [84][85][86][88][92][91][93]. Using Western blot analysis, several studies showed significant decreased platelet ADAM-10 activity associated to a heightened activity of the active BACE-1 forms and, in some cases, reduced level of α-APPs (reduced concomitantly in CSF) [94][84][85][86][88]. Platelet ADAM-10 negatively correlated with the severity of cognitive impairment [94]. Recently, the decreased platelet ADAM-10 activity was associated to lower platelet presenilin-1 (PSEN1) levels in AD patients compared to age-matched controls. This association did not emerge in leukocytes suggesting probably that platelets represent a more reliable peripheral matrix than leukocytes to study the APP processing system [95].
β-Secretase activity was also measured with a different method (Calbiochem, β-secretase Substrate I) that confirmed an increased β-secretase activity in MCI and AD subjects compared to age-matched control group [93][96][97].
Interestingly, in a two-year follow-up study, baseline platelet membrane β-secretase activity was investigated in 97 MCI subjects and 85 controls. At T0, platelet β-secretase activity did not differ significantly between groups but, at the final endpoint, total enzyme activity tended to be 10% higher in MCI participants. β-secretase activity was measured using a commercially available fluorogenic substrate, Sigma A1472 [98]. This study was the first to investigate the assay signal measuring activity in the presence and absence of two BACE inhibitors. Although this method was imperfect because of the lack of inhibitor specificity, it could provide a more specific measure of enzyme activity.
Differently, other studies did not replicate these findings and, in some cases, showed contrasting results. Gorham et al. (2010) analyzed the processing enzymes in a Swedish population of 20 AD patients, 6 MCI patients and 30 healthy controls. They did not find any significative differences among groups. However, they observed an inverse correlation between plasma triacylglycerol (TAG) levels and the secretase ratio [99]. A cross-sectional exhibited decreased levels of several BACE-1 isoforms in the AD sample compared to controls [100][101].
3. Conclusions
The studies that have so far dealt with the alterations in the processing of the APP, both those concerning the investigation of the APP ratio and those that analyze the activity degree of the amylodogenic and non-amylodogenic pathway, converges in an almost unitary way in affirming that subjects with AD show changes in APP processing system compared to healthy age-matched controls. Often, these alterations correlate with cognitive impairment severity and with functional autonomy. Furthermore, these alterations do not only occur in parallel to the cognitive decay process but, in some cases, they are detectable in the preclinical stages (aMCI and MCI), suggesting their use as a potential early ante-mortem marker AD clinical diagnosis. To support these findings and to promote the potential use of these biomarkers in the therapeutic field, there are several clinical trials that tested the use of the acetylcholine (ACh) esterase inhibitor, Donepezil (5 mg/day) and Galantamine, in AD patients [78][81][102][103][104][105]. Subjects with AD, comparing to controls, showed an increase in platelet APPr and in MMSE score. The modification of APPr was influenced by ApoE genotype as the non-ε4 carriers showed a higher APPr recovery. Furthermore, some authors stated that AChEIs treatment rescues impaired APP metabolism increasing significantly ADAM10 levels, α-secretase activity and reducing β-secretase cleavage [104][105]. Similarly, AD patients treated for six weeks with anticholesterol drugs (Statin or Niacin) showed an increased APPr therefore limiting Aβ secretion from platelets [106][107][108].
Alteration of the APP processing system in AD patients is beyond doubt, but the exact cause of these changes is still controverting. It has long been known that APP is found in megakaryocytes as well as in the platelet α-granules in relatively high concentrations and it is released in plasma during platelet activation [26][70][77][79][80][109].
Rosenberg et al. in 1997 were the first to highlight the possibility of a platelet activation in AD patients related to altered APP processing [80]. In the following years, several research groups confirmed the presence of an aberrant and chronic pre-activation of platelets that can eventually contribute towards atherothrombosis, CAA, and progression of AD [110].
The declining ratio of APP isoforms in platelets may result from increased release of the 130 kDa isoforms upon platelet degranulation [71][111]. Blood platelets could be an undoubted additional source of Aβ in the brain, especially in Aβ accumulation in sub-endothelium of blood vessels, since Aβ is stored in α-granules and directly released by platelet [23][109] or cleaved from platelet APP. It is cleaved after release by platelet BACE-1 or by the endothelial cells of brain blood vessels [112].
The activated platelets in AD patients retain greater amounts of APP, show more platelet adhesion and thrombus formation. These characteristics lead to a greater possibility for the platelets to aggregate in clots releasing massive quantity of APP and Aβ [73][113]. Vessel damage is a natural cause of platelet activation and degranulation. Aβ protein accumulated around blood vessels forms the characteristic fiche of Alzheimer’s amyloid angiopathy [114][115]. Aβ have been shown to activate platelets and act as positive modulators. This molecule induces platelet aggregation and, in the meanwhile, increases significantly the responses to low levels of physiological agonists. This would trigger a circuit that lead to a noticeable increase in platelet aggregability with the consequent risk of an unwanted hemostatic response and clot formation leading to thrombosis.
Furthermore, platelet derived Aβ passes through the BBB by the mechanism of binding to apolipoproteins. Advanced glycation end products (RAGE) receptor, the low-density lipoprotein receptor-related protein 1 (LRP1), the P-glycoprotein (also known as ABCB1) and the BCRP (also known as ABCG2) are involved in the influx-efflux transport of Aβ from the brain [116][117][118][119][120]. Both brain- and blood-derived Aβ peptide may overwhelm the capacity of the existing clearance system. This hypothesis is in agreement with the recent discovery of the glymphatic system, which suggests an alternative way of perivascular clearance of Aβ without going back into the blood [121][121][122].
Conditions that can potentially burden on the integrity of the cell membrane of brain endothelial cells, that form a system of tight junctions in order to regulate communication between the brain and circulating blood factors, like being carriers of ApoE allele ε 4, impact cerebral and vascular systems making prone to the onset of Alzheimer disease, cardiovascular disorders and stroke (Figure 2).
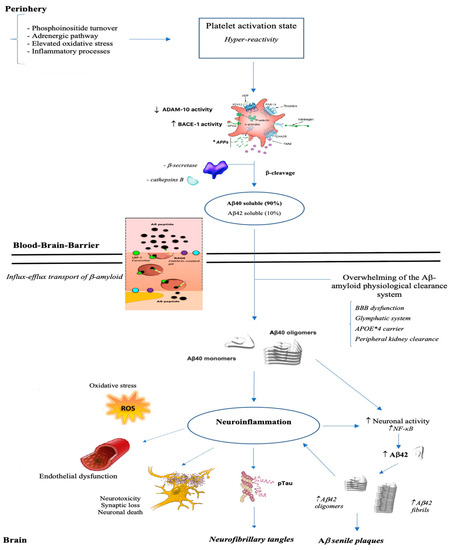
Figure 2. From brain to periphery: a state of platelet hyper-reactivity implicates an increase in the production of Aβ which, once crossed the blood-brain barrier, polymerizes into aggregates, deposits and triggers a neuroinflammatory process.
Finally, we can conclude that platelets represent a promising peripheral model for detecting and understanding the molecular changes related to the onset of AD, while providing crucial data necessary towards the development of an effective diagnostic tool and/or, above all, towards the elaboration of therapeutic solutions. Despite the massive presence of data, at the current state of the art, none of the individual markers described is powerful enough to meet the required levels of sensitivity and specificity for the routine diagnosis of AD, it could be useful to exploit these candidate biomarkers simultaneously.
References
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344.
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol. 2013, 12, 357–367.
- Reiman, E.M.; Quiroz, Y.T.; Fleisher, A.S.; Chen, K.; Velez-Pardo, C.; Jimenez-Del-Rio, M.; Fagan, A.M.; Shah, A.R.; Alvarez, S.; Arbelaez, A.; et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: A case-control study. Lancet Neurol. 2012, 11, 1048–1056.
- Jack, C.R., Jr.; Lowe, V.J.; Weigand, S.D.; Wiste, H.J.; Senjem, M.L.; Knopman, D.S.; Shiung, M.M.; Gunter, J.L.; Boeve, B.F.; Kemp, B.J.; et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: Implications for sequence of pathological events in Alzheimer’s disease. Brain 2009, 132, 1355–1365.
- Gordon, B.A.; Blazey, T.M.; Su, Y.; Hari-Raj, A.; Dincer, A.; Flores, S.; Christensen, J.; McDade, E.; Wang, G.; Xiong, C.; et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: A longitudinal study. Lancet Neurol. 2018, 17, 241–250.
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Dominantly Inherited Alzheimer, N., Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804.
- Thal, D.R.; Del Tredici, K.; Ludolph, A.C.; Hoozemans, J.J.; Rozemuller, A.J.; Braak, H.; Knippschild, U. Stages of granulovacuolar degeneration: Their relation to Alzheimer’s disease and chronic stress response. Acta Neuropathol. 2011, 122, 577–589.
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969.
- Den Haan, J.; Morrema, T.H.J.; Verbraak, F.D.; de Boer, J.F.; Scheltens, P.; Rozemuller, A.J.; Bergen, A.A.B.; Bouwman, F.H.; Hoozemans, J.J. Amyloid-beta and phosphorylated tau in post-mortem Alzheimer’s disease retinas. Acta Neuropathol. Commun. 2018, 6, 147.
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547.
- Villemagne, V.L.; Rowe, C.C. Amyloid imaging. Int. Psychogeriatr. 2011, 23 (Suppl. 2), S41–S49.
- Villemagne, V.L.; Ong, K.; Mulligan, R.S.; Holl, G.; Pejoska, S.; Jones, G.; O’Keefe, G.; Ackerman, U.; Tochon-Danguy, H.; Chan, J.G.; et al. Amyloid imaging with (18)F-florbetaben in Alzheimer disease and other dementias. J. Nucl. Med. 2011, 52, 1210–1217.
- Villemagne, V.L.; O’Keefe, G.; Mulligan, R.S.; Rowe, C.C. Quantitative approaches to amyloid imaging. Methods Mol. Biol. 2011, 680, 201–225.
- Blennow, K.; Hampel, H.; Weiner, M.; Zetterberg, H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 131–144.
- Hampel, H.; Buerger, K.; Zinkowski, R.; Teipel, S.J.; Goernitz, A.; Andreasen, N.; Sjoegren, M.; DeBernardis, J.; Kerkman, D.; Ishiguro, K.; et al. Measurement of phosphorylated tau epitopes in the differential diagnosis of Alzheimer disease: A comparative cerebrospinal fluid study. Arch. Gen. Psychiatry 2004, 61, 95–102.
- Mattsson, N.; Zetterberg, H.; Hansson, O.; Andreasen, N.; Parnetti, L.; Jonsson, M.; Herukka, S.K.; van der Flier, W.M.; Blankenstein, M.A.; Ewers, M.; et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA 2009, 302, 385–393.
- Mattsson, N.; Zetterberg, H. Future screening for incipient Alzheimer’s disease--the influence of prevalence on test performance. Eur. Neurol. 2009, 62, 200–203.
- Blennow, K. A Review of Fluid Biomarkers for Alzheimer’s Disease: Moving from CSF to Blood. Neurology 2017, 6 (Suppl. 1), 15–24.
- Henriksen, K.; O’Bryant, S.E.; Hampel, H.; Trojanowski, J.Q.; Montine, T.J.; Jeromin, A.; Blennow, K.; Lonneborg, A.; Wyss-Coray, T.; Soares, H.; et al. The future of blood-based biomarkers for Alzheimer’s disease. Alzheimers Dement. 2014, 10, 115–131.
- Galasko, D.; Golde, T.E. Biomarkers for Alzheimer’s disease in plasma, serum and blood—Conceptual and practical problems. Alzheimers Res. 2013, 5, 10.
- Olsson, B.; Lautner, R.; Andreasson, U.; Ohrfelt, A.; Portelius, E.; Bjerke, M.; Holtta, M.; Rosen, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684.
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Dore, V.; Fowler, C.; Li, Q.X.; Martins, R.; Rowe, C.; et al. High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature 2018, 554, 249–254.
- Chen, M.; Inestrosa, N.C.; Ross, G.S.; Fernandez, H.L. Platelets are the primary source of amyloid beta-peptide in human blood. Biochem. Biophys. Res. Commun. 1995, 213, 96–103.
- Reed, G.L. Platelet secretory mechanisms. Semin. Thromb. Hemost. 2004, 30, 441–450.
- Smith, C.C. Stimulated release of the beta-amyloid protein of Alzheimer’s disease by normal human platelets. Neurosci. Lett. 1997, 235, 157–159.
- Di Luca, M.C.F.; Pastorino, L.; Borroni, B.; Padovani, A.; Cattabeni, F. Platelets as a peripheral district where to study pathogenetic mechanisms of alzheimer disease: The case of amyloid precursor protein. Eur. J. Pharm. 2000, 405, 277–283.
- Bayer, T.A.; Cappai, R.; Masters, C.L.; Beyreuther, K.; Multhaup, G. It all sticks together—The APP-related family of proteins and Alzheimer’s disease. Mol. Psychiatry 1999, 4, 524–528.
- Skovronsky, D.M.; Lee, V.M.; Pratico, D. Amyloid precursor protein and amyloid beta peptide in human platelets. Role of cyclooxygenase and protein kinase C. J. Biol. Chem. 2001, 276, 17036–17043.
- Racchi, M.; Govoni, S. The pharmacology of amyloid precursor protein processing. Exp. Gerontol. 2003, 38, 145–157.
- Haass, C. Take five--BACE and the gamma-secretase quartet conduct Alzheimer’s amyloid beta-peptide generation. EMBO J. 2004, 23, 483–488.
- Zainaghi, I.A.; Forlenza, O.V.; Gattaz, W.F. Abnormal APP processing in platelets of patients with Alzheimer’s disease: Correlations with membrane fluidity and cognitive decline. Psychopharmacology 2007, 192, 547–553.
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Muller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736.
- Muller, U.C.; Pietrzik, C.U.; Deller, T. The physiological functions of the beta-amyloid precursor protein APP. Exp. Brain Res. 2012, 217, 325–329.
- Visconte, C.; Canino, J.; Guidetti, G.F.; Zara, M.; Seppi, C.; Abubaker, A.A.; Pula, G.; Torti, M.; Canobbio, I. Amyloid precursor protein is required for in vitro platelet adhesion to amyloid peptides and potentiation of thrombus formation. Cell Signal 2018, 52, 95–102.
- Silva, J.V.; Yoon, S.; Domingues, S.; Guimaraes, S.; Goltsev, A.V.; da Cruz, E.S.E.F.; Mendes, J.F.; da Cruz, E.S.O.A.; Fardilha, M. Amyloid precursor protein interaction network in human testis: Sentinel proteins for male reproduction. BMC Bioinform. 2015, 16, 12.
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608.
- Sloane, J.A.; Pietropaolo, M.F.; Rosene, D.L.; Moss, M.B.; Peters, A.; Kemper, T.; Abraham, C.R. Lack of correlation between plaque burden and cognition in the aged monkey. Acta Neuropathol. 1997, 94, 471–478.
- Sengupta, U.; Nilson, A.N.; Kayed, R. The Role of Amyloid-beta Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 2016, 6, 42–49.
- Jeong, S. Molecular and Cellular Basis of Neurodegeneration in Alzheimer’s Disease. Mol. Cells 2017, 40, 613–620.
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Cellular Receptors of Amyloid beta Oligomers (AbetaOs) in Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 1884.
- Monning, U.; Konig, G.; Banati, R.B.; Mechler, H.; Czech, C.; Gehrmann, J.; Schreiter-Gasser, U.; Masters, C.L.; Beyreuther, K. Alzheimer beta A4-amyloid protein precursor in immunocompetent cells. J. Biol. Chem. 1992, 267, 23950–23956.
- Golde, T.E.; Estus, S.; Usiak, M.; Younkin, L.H.; Younkin, S.G. Expression of beta amyloid protein precursor mRNAs: Recognition of a novel alternatively spliced form and quantitation in Alzheimer’s disease using PCR. Neuron 1990, 4, 253–267.
- Krieger, M.A.; Salles, J.M.; Almeida, E.; Linss, J.; Bonaldo, M.C.; Goldenberg, S. Expression and polymorphism of a Trypanosoma cruzi gene encoding a cytoplasmic repetitive antigen. Exp. Parasitol. 1990, 70, 247–254.
- Li, Q.X.; Berndt, M.C.; Bush, A.I.; Rumble, B.; Mackenzie, I.; Friedhuber, A.; Beyreuther, K.; Masters, C.L. Membrane-associated forms of the beta A4 amyloid protein precursor of Alzheimer’s disease in human platelet and brain: Surface expression on the activated human platelet. Blood 1994, 84, 133–142.
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204.
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619.
- Buxbaum, J.D.; Liu, K.N.; Luo, Y.; Slack, J.L.; Stocking, K.L.; Peschon, J.J.; Johnson, R.S.; Castner, B.J.; Cerretti, D.P.; Black, R.A. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J. Biol. Chem. 1998, 273, 27765–27767.
- Lammich, S.; Kojro, E.; Postina, R.; Gilbert, S.; Pfeiffer, R.; Jasionowski, M.; Haass, C.; Fahrenholz, F. Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. USA 1999, 96, 3922–3927.
- Chang, C.; Werb, Z. The many faces of metalloproteases: Cell growth, invasion, angiogenesis and metastasis. Trends Cell Biol. 2001, 11, S37–S43.
- Cole, S.L.; Vassar, R. BACE1 structure and function in health and Alzheimer’s disease. Curr. Alzheimer Res. 2008, 5, 100–120.
- Seals, D.F.; Courtneidge, S.A. The ADAMs family of metalloproteases: Multidomain proteins with multiple functions. Genes Dev. 2003, 17, 7–30.
- Vingtdeux, V.; Marambaud, P. Identification and biology of alpha-secretase. J. Neurochem. 2012, 120 (Suppl. 1), 34–45.
- Sobol, A.; Galluzzo, P.; Liang, S.; Rambo, B.; Skucha, S.; Weber, M.J.; Alani, S.; Bocchetta, M. Amyloid precursor protein (APP) affects global protein synthesis in dividing human cells. J. Cell Physiol. 2015, 230, 1064–1074.
- Kucheryavykh, L.Y.; Davila-Rodriguez, J.; Rivera-Aponte, D.E.; Zueva, L.V.; Washington, A.V.; Sanabria, P.; Inyushin, M.Y. Platelets are responsible for the accumulation of beta-amyloid in blood clots inside and around blood vessels in mouse brain after thrombosis. Brain Res. Bull 2017, 128, 98–105.
- Gandy, S. Lifelong management of amyloid-beta metabolism to prevent Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 864–866.
- Venugopal, C.; Demos, C.M.; Rao, K.S.; Pappolla, M.A.; Sambamurti, K. Beta-secretase: Structure, function, and evolution. CNS Neurol. Disord. Drug Targets 2008, 7, 278–294.
- Vidal, R.; Ghiso, J.; Wisniewski, T.; Frangione, B. Alzheimer’s presenilin 1 gene expression in platelets and megakaryocytes. Identification of a novel splice variant. FEBS Lett. 1996, 393, 19–23.
- Cole, S.L.; Vassar, R. The Alzheimer’s disease beta-secretase enzyme, BACE1. Mol. Neurodegener. 2007, 2, 22.
- De Strooper, B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007, 8, 141–146.
- Ganjei, J.K. Targeting amyloid precursor protein secretases: Alzheimer’s disease and beyond. Drug News Perspect. 2010, 23, 573–584.
- Jain, P.; Wadhwa, P.K.; Rohilla, S.; Jadhav, H.R. Rational design, synthesis and in vitro evaluation of allylidene hydrazinecarboximidamide derivatives as BACE-1 inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 33–37.
- Takami, M.; Nagashima, Y.; Sano, Y.; Ishihara, S.; Morishima-Kawashima, M.; Funamoto, S.; Ihara, Y. gamma-Secretase: Successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J. Neurosci. 2009, 29, 13042–13052.
- Kakuda, N.; Shoji, M.; Arai, H.; Furukawa, K.; Ikeuchi, T.; Akazawa, K.; Takami, M.; Hatsuta, H.; Murayama, S.; Hashimoto, Y.; et al. Japanese Alzheimer’s Disease Neuroimaging, I., Altered gamma-secretase activity in mild cognitive impairment and Alzheimer’s disease. EMBO Mol. Med. 2012, 4, 344–352.
- Takami, M.; Funamoto, S. gamma-Secretase-Dependent Proteolysis of Transmembrane Domain of Amyloid Precursor Protein: Successive Tri- and Tetrapeptide Release in Amyloid beta-Protein Production. Int. J. Alzheimers Dis. 2012, 2012, 591392.
- Smirnov, A.; Trupp, A.; Henkel, A.W.; Bloch, E.; Reulbach, U.; Lewczuk, P.; Riggert, J.; Kornhuber, J.; Wiltfang, J. Differential processing and secretion of Abeta peptides and sAPPalpha in human platelets is regulated by thrombin and prostaglandine 2. Neurobiol. Aging 2009, 30, 1552–1562.
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890.
- Glenner, G.G.; Wong, C.W.; Quaranta, V.; Eanes, E.D. The amyloid deposits in Alzheimer’s disease: Their nature and pathogenesis. Appl. Pathol. 1984, 2, 357–369.
- Masters, C.L.; Multhaup, G.; Simms, G.; Pottgiesser, J.; Martins, R.N.; Beyreuther, K. Neuronal origin of a cerebral amyloid: Neurofibrillary tangles of Alzheimer’s disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J. 1985, 4, 2757–2763.
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249.
- Bush, A.I.; Martins, R.N.; Rumble, B.; Moir, R.; Fuller, S.; Milward, E.; Currie, J.; Ames, D.; Weidemann, A.; Fischer, P.; et al. The amyloid precursor protein of Alzheimer’s disease is released by human platelets. J. Biol. Chem. 1990, 265, 15977–15983.
- Bush, A.I.; Whyte, S.; Thomas, L.D.; Williamson, T.G.; Van Tiggelen, C.J.; Currie, J.; Small, D.H.; Moir, R.D.; Li, Q.X.; Rumble, B.; et al. An abnormality of plasma amyloid protein precursor in Alzheimer’s disease. Ann. Neurol. 1992, 32, 57–65.
- Davies, T.A.; Fine, R.E.; Johnson, R.J.; Levesque, C.A.; Rathbun, W.H.; Seetoo, K.F.; Smith, S.J.; Strohmeier, G.; Volicer, L.; Delva, L.; et al. Non-age related differences in thrombin responses by platelets from male patients with advanced Alzheimer’s disease. Biochem. Biophys. Res. Commun. 1993, 194, 537–543.
- Davies, T.A.; Long, H.J.; Sgro, K.; Rathbun, W.H.; McMenamin, M.E.; Seetoo, K.; Tibbles, H.; Billingslea, A.M.; Fine, R.E.; Fishman, J.B.; et al. Activated Alzheimer disease platelets retain more beta amyloid precursor protein. Neurobiol. Aging 1997, 18, 147–153.
- Davies, T.A.; Long, H.J.; Tibbles, H.E.; Sgro, K.R.; Wells, J.M.; Rathbun, W.H.; Seetoo, K.F.; McMenamin, M.E.; Smith, S.J.; Feldman, R.G.; et al. Moderate and advanced Alzheimer’s patients exhibit platelet activation differences. Neurobiol. Aging 1997, 18, 155–162.
- Di Luca, M.; Pastorino, L.; Cattabeni, F.; Zanardi, R.; Scarone, S.; Racagni, G.; Smeraldi, E.; Perez, J. Abnormal pattern of platelet APP isoforms in Alzheimer disease and Down syndrome. Arch. Neurol. 1996, 53, 1162–1166.
- Di Luca, M.; Pastorino, L.; Bianchetti, A.; Perez, J.; Vignolo, L.A.; Lenzi, G.L.; Trabucchi, M.; Cattabeni, F.; Padovani, A. Differential level of platelet amyloid beta precursor protein isoforms: An early marker for Alzheimer disease. Arch. Neurol. 1998, 55, 1195–1200.
- Padovani, A.; Pastorino, L.; Borroni, B.; Colciaghi, F.; Rozzini, L.; Monastero, R.; Perez, J.; Pettenati, C.; Mussi, M.; Parrinello, G.; et al. Amyloid precursor protein in platelets: A peripheral marker for the diagnosis of sporadic AD. Neurology 2001, 57, 2243–2248.
- Borroni, B.; Colciaghi, F.; Pastorino, L.; Archetti, S.; Corsini, P.; Cattabeni, F.; Di Luca, M.; Padovani, A. ApoE genotype influences the biological effect of donepezil on APP metabolism in Alzheimer disease: Evidence from a peripheral model. Eur. Neuropsychopharmacol. 2002, 12, 195–200.
- Baskin, F.; Rosenberg, R.N.; Iyer, L.; Hynan, L.; Cullum, C.M. Platelet APP isoform ratios correlate with declining cognition in AD. Neurology 2000, 54, 1907–1909.
- Rosenberg, R.N.; Baskin, F.; Fosmire, J.A.; Risser, R.; Adams, P.; Svetlik, D.; Honig, L.S.; Cullum, C.M.; Weiner, M.F. Altered amyloid protein processing in platelets of patients with Alzheimer disease. Arch. Neurol. 1997, 54, 139–144.
- Borroni, B.; Colciaghi, F.; Pastorino, L.; Pettenati, C.; Cottini, E.; Rozzini, L.; Monastero, R.; Lenzi, G.L.; Cattabeni, F.; Di Luca, M.; et al. Amyloid precursor protein in platelets of patients with Alzheimer disease: Effect of acetylcholinesterase inhibitor treatment. Arch. Neurol. 2001, 58, 442–446.
- Sanchez-Gonzalez, V.J.; Ortiz, G.G.; Gallegos-Arreola, P.; Macias-Islas, M.A.; Arias-Merino, E.D.; Loera-Castaneda, V.; Martinez-Cano, E.; Velazquez-Brizuela, I.E.; Rosales-Corral, S.A.; Curiel-Ortega, C.R.; et al. Altered beta-amyloid precursor protein isoforms in Mexican Alzheimer’s Disease patients. Dis. Markers 2006, 22, 119–125.
- Shi, Y.; Gu, L.; Wang, Q.; Gao, L.; Zhu, J.; Lu, X.; Zhou, F.; Zhu, D.; Zhang, H.; Xie, C.; et al. Platelet Amyloid-beta Protein Precursor (AbetaPP) Ratio and Phosphorylated Tau as Promising Indicators for Early Alzheimer’s Disease. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 75, 664–670.
- Colciaghi, F.; Marcello, E.; Borroni, B.; Zimmermann, M.; Caltagirone, C.; Cattabeni, F.; Padovani, A.; Di Luca, M. Platelet APP, ADAM 10 and BACE alterations in the early stages of Alzheimer disease. Neurology 2004, 62, 498–501.
- Colciaghi, F.; Borroni, B.; Pastorino, L.; Marcello, E.; Zimmermann, M.; Cattabeni, F.; Padovani, A.; Di Luca, M. [alpha]-Secretase ADAM10 as well as [alpha]APPs is reduced in platelets and CSF of Alzheimer disease patients. Mol. Med. 2002, 8, 67–74.
- Tang, K.; Hynan, L.S.; Baskin, F.; Rosenberg, R.N. Platelet amyloid precursor protein processing: A bio-marker for Alzheimer’s disease. J. Neurol. Sci. 2006, 240, 53–58.
- Borroni, B.; Perani, D.; Broli, M.; Colciaghi, F.; Garibotto, V.; Paghera, B.; Agosti, C.; Giubbini, R.; Di Luca, M.; Padovani, A. Pre-clinical diagnosis of Alzheimer disease combining platelet amyloid precursor protein ratio and rCBF spect analysis. J. Neurol. 2005, 252, 1359–1362.
- Di Luca, M.; Grossi, E.; Borroni, B.; Zimmermann, M.; Marcello, E.; Colciaghi, F.; Gardoni, F.; Intraligi, M.; Padovani, A.; Buscema, M. Artificial neural networks allow the use of simultaneous measurements of Alzheimer disease markers for early detection of the disease. J. Transl. Med. 2005, 3, 30.
- Vignini, A.; Sartini, D.; Morganti, S.; Nanetti, L.; Luzzi, S.; Provinciali, L.; Mazzanti, L.; Emanuelli, M. Platelet amyloid precursor protein isoform expression in Alzheimer’s disease: Evidence for peripheral marker. Int. J. Immunopathol. Pharm. 2011, 24, 529–534.
- Vignini, A.; Morganti, S.; Salvolini, E.; Sartini, D.; Luzzi, S.; Fiorini, R.; Provinciali, L.; Di Primio, R.; Mazzanti, L.; Emanuelli, M. Amyloid precursor protein expression is enhanced in human platelets from subjects with Alzheimer’s disease and frontotemporal lobar degeneration: A real-time PCR study. Exp. Gerontol. 2013, 48, 1505–1508.
- Mukaetova-Ladinska, E.B.; Abdel-All, Z.; Dodds, S.; Andrade, J.; Alves da Silva, J.; Kalaria, R.N.; O’Brien, J.T. Platelet immunoglobulin and amyloid precursor protein as potential peripheral biomarkers for Alzheimer’s disease: Findings from a pilot study. Age Ageing 2012, 41, 408–412.
- Liu, H.C.; Wang, H.C.; Ko, S.Y.; Wang, P.N.; Chi, C.W.; Hong, C.J.; Lin, K.N.; Liu, T.Y. Correlation between platelet amyloid precursor protein isoform ratio and cognition in Alzheimer’s disease. J. Alzheimers Dis. 2007, 11, 77–84.
- Liu, W.W.; Todd, S.; Craig, D.; Passmore, A.P.; Coulson, D.T.; Murphy, S.; Irvine, G.B.; Johnston, J.A. Elevated platelet beta-secretase activity in mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 2007, 24, 464–468.
- Manzine, P.R.; de Franca Bram, J.M.; Barham, E.J.; do Vale Fde, A.; Selistre-de-Araujo, H.S.; Cominetti, M.R.; Iost Pavarini, S.C. ADAM10 as a biomarker for Alzheimer’s disease: A study with Brazilian elderly. Dement Geriatr. Cogn. Disord. 2013, 35, 58–66.
- Bram, J.M.F.; Talib, L.L.; Joaquim, H.P.G.; Sarno, T.A.; Gattaz, W.F.; Forlenza, O.V. Protein levels of ADAM10, BACE1, and PSEN1 in platelets and leukocytes of Alzheimer’s disease patients. Eur. Arch. Psychiatry Clin. Neurosci. 2019, 269, 963–972.
- Bermejo-Bescos, P.; Martin-Aragon, S.; Jimenez-Aliaga, K.; Benedi, J.; Felici, E.; Gil, P.; Ribera, J.M.; Villar, A.M. Processing of the platelet amyloid precursor protein in the mild cognitive impairment (MCI). Neurochem. Res. 2013, 38, 1415–1423.
- Johnston, J.A.; Liu, W.W.; Coulson, D.T.; Todd, S.; Murphy, S.; Brennan, S.; Foy, C.J.; Craig, D.; Irvine, G.B.; Passmore, A.P. Platelet beta-secretase activity is increased in Alzheimer’s disease. Neurobiol. Aging 2008, 29, 661–668.
- McGuinness, B.; Fuchs, M.; Barrett, S.L.; Passmore, A.P.; Johnston, J.A. Platelet Membrane beta-Secretase Activity in Mild Cognitive Impairment and Conversion to Dementia: A Longitudinal Study. J. Alzheimers Dis. 2016, 49, 1095–1103.
- Gorham, P.; Bark, N.; Bjorkhem, I.; Meaney, S.; Crisby, M. Platelet alpha-and beta- secretase activities are not significantly affected by dementia or mild cognitive impairment in Swedish patients. Curr. Alzheimer Res. 2010, 7, 134–139.
- Gonzales, A.; Decourt, B.; Walker, A.; Condjella, R.; Nural, H.; Sabbagh, M.N. Development of a specific ELISA to measure BACE1 levels in human tissues. J. Neurosci. Methods 2011, 202, 70–76.
- Decourt, B.; Walker, A.; Gonzales, A.; Malek-Ahmadi, M.; Liesback, C.; Davis, K.J.; Belden, C.M.; Jacobson, S.A.; Sabbagh, M.N. Can platelet BACE1 levels be used as a biomarker for Alzheimer’s disease? Proof-of-concept study. Platelets 2013, 24, 235–238.
- Mukaetova-Ladinska, E.B.; Abdel-All, Z.; Andrade, J.; McNally, R.J.; James, P.W.; Kalaria, R.N.; O’Brien, J.T. Increase in platelet immunoglobulin in Alzheimer’s disease is normalised following cholinesterase inhibitor treatment: Preliminary results. J. Alzheimers Dis. 2012, 32, 431–436.
- Liu, H.C.; Chi, C.W.; Ko, S.Y.; Wang, H.C.; Hong, C.J.; Lin, K.N.; Wang, P.N.; Liu, T.Y. Cholinesterase inhibitor affects the amyloid precursor protein isoforms in patients with Alzheimer’s disease. Dement Geriatr. Cogn. Disord. 2005, 19, 345–348.
- Sarno, T.A.; Talib, L.L.; Joaquim, H.P.; Bram, J.M.; Gattaz, W.F.; Forlenza, O.V. Protein Expression of BACE1 is Downregulated by Donepezil in Alzheimer’s Disease Platelets. J. Alzheimers Dis. 2017, 55, 1445–1451.
- Zimmermann, M.; Borroni, B.; Cattabeni, F.; Padovani, A.; Di Luca, M. Cholinesterase inhibitors influence APP metabolism in Alzheimer disease patients. Neurobiol. Dis. 2005, 19, 237–242.
- Borroni, B.; Colciaghi, F.; Lenzi, G.L.; Caimi, L.; Cattabeni, F.; Di Luca, M.; Padovani, A. High cholesterol affects platelet APP processing in controls and in AD patients. Neurobiol. Aging 2003, 24, 631–636.
- Baskin, F.; Rosenberg, R.N.; Fang, X.; Hynan, L.S.; Moore, C.B.; Weiner, M.; Vega, G.L. Correlation of statin-increased platelet APP ratios and reduced blood lipids in AD patients. Neurology 2003, 60, 2006–2007.
- Grimm, M.O.; Mett, J.; Grimm, H.S.; Hartmann, T. APP Function and Lipids: A Bidirectional Link. Front. Mol. Neurosci. 2017, 10, 63.
- Van Nostrand, W.E.; Schmaier, A.H.; Farrow, J.S.; Cunningham, D.D. Protease nexin-II (amyloid beta-protein precursor): A platelet alpha-granule protein. Science 1990, 248, 745–748.
- Sevush, S.; Jy, W.; Horstman, L.L.; Mao, W.W.; Kolodny, L.; Ahn, Y.S. Platelet activation in Alzheimer disease. Arch. Neurol. 1998, 55, 530–536.
- Youmans, K.L.; Tai, L.M.; Kanekiyo, T.; Stine, W.B., Jr.; Michon, S.C.; Nwabuisi-Heath, E.; Manelli, A.M.; Fu, Y.; Riordan, S.; Eimer, W.A.; et al. Intraneuronal Abeta detection in 5xFAD mice by a new Abeta-specific antibody. Mol. Neurodegener. 2012, 7, 8.
- Davies, T.A.; Billingslea, A.M.; Long, H.J.; Tibbles, H.; Wells, J.M.; Eisenhauer, P.B.; Smith, S.J.; Cribbs, D.H.; Fine, R.E.; Simons, E.R. Brain endothelial cell enzymes cleave platelet-retained amyloid precursor protein. J. Lab. Clin. Med. 1998, 132, 341–350.
- Canobbio, I.; Visconte, C.; Oliviero, B.; Guidetti, G.; Zara, M.; Pula, G.; Torti, M. Increased platelet adhesion and thrombus formation in a mouse model of Alzheimer’s disease. Cell Signal 2016, 28, 1863–1871.
- Weller, R.O.; Nicoll, J.A. Cerebral amyloid angiopathy: Pathogenesis and effects on the ageing and Alzheimer brain. Neurol. Res. 2003, 25, 611–616.
- Vinters, H.V. Cerebral amyloid angiopathy. A critical review. Stroke 1987, 18, 311–324.
- Davies, T.A.; Long, H.J.; Eisenhauer, P.B.; Hastey, R.; Cribbs, D.H.; Fine, R.E.; Simons, E.R. Beta amyloid fragments derived from activated platelets deposit in cerebrovascular endothelium: Usage of a novel blood brain barrier endothelial cell model system. Amyloid 2000, 7, 153–165.
- Deane, R.; Bell, R.D.; Sagare, A.; Zlokovic, B.V. Clearance of amyloid-beta peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 16–30.
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913.
- Shayo, M.; McLay, R.N.; Kastin, A.J.; Banks, W.A. The putative blood-brain barrier transporter for the beta-amyloid binding protein apolipoprotein j is saturated at physiological concentrations. Life Sci. 1997, 60, PL115–PL118.
- Zhang, W.; Xiong, H.; Callaghan, D.; Liu, H.; Jones, A.; Pei, K.; Fatehi, D.; Brunette, E.; Stanimirovic, D. Blood-brain barrier transport of amyloid beta peptides in efflux pump knock-out animals evaluated by in vivo optical imaging. Fluids Barriers CNS 2013, 10, 13.
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341, Corrigendum in 2016, 533, 278.
- Weller, R.O.; Subash, M.; Preston, S.D.; Mazanti, I.; Carare, R.O. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 2008, 18, 253–266.
More
Information
Subjects:
Neurosciences
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
671
Revisions:
2 times
(View History)
Update Date:
26 Aug 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No