Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Annalisa Noce | + 2482 word(s) | 2482 | 2021-08-24 11:46:47 | | | |
| 2 | Rita Xu | Meta information modification | 2482 | 2021-08-25 04:09:43 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Noce, A. Endothelial Dysfunction in CKD. Encyclopedia. Available online: https://encyclopedia.pub/entry/13514 (accessed on 07 February 2026).
Noce A. Endothelial Dysfunction in CKD. Encyclopedia. Available at: https://encyclopedia.pub/entry/13514. Accessed February 07, 2026.
Noce, Annalisa. "Endothelial Dysfunction in CKD" Encyclopedia, https://encyclopedia.pub/entry/13514 (accessed February 07, 2026).
Noce, A. (2021, August 24). Endothelial Dysfunction in CKD. In Encyclopedia. https://encyclopedia.pub/entry/13514
Noce, Annalisa. "Endothelial Dysfunction in CKD." Encyclopedia. Web. 24 August, 2021.
Copy Citation
Chronic kidney disease (CKD) represents a world-wide public health problem. Inflammation, endothelial dysfunction (ED) and vascular calcifications are clinical features of CKD patients that increase cardiovascular (CV) mortality. CKD-related CV disease pathogenic mechanisms are not only associated with traditional factors such as arterial hypertension and dyslipidemia, but also with ED, oxidative stress and low-grade inflammation.
cardiovascular disease
chronic kidney disease
endothelial dysfunction
endothelium
1. Introduction
Chronic kidney disease (CKD) is an increasing health problem both socially and economically, worldwide [1]. Patients in renal replacement therapy (RRT) have high mortality, mainly related to cardiovascular diseases (CVDs) [2][3]. The enhanced incidence of cardiovascular (CV) events is closely related to the state of chronic inflammation, typical of CKD, resulting in the acceleration of ageing phenomena. In fact, CKD is currently considered to be an early ageing model [4].
An important epidemiological study demonstrated that hemodialysis (HD) patients aged 25–35 years have a higher CV-mortality rate than subjects over 85 years of the general population. This phenomenon cannot be explained by examining the traditional “modifiable” and the “non modifiable” CV risk factors such as gender, age, tobacco habit, etc. [5][6][7][8]. For this reason, CV risk factors related to uremia need to be considered. These specific CV risk factors are multiple: (i) alterations of calcium-phosphorus metabolism, (ii) hyperhomocysteinemia, (iii) endothelial dysfunction (ED), (iv) oxidative stress (OS), (v) chronic inflammation, (vi) increased asymmetric dimethylarginine (ADMA), (vii) albuminuria, (viii) malnutrition and (ix) uremic sarcopenia (Figure 1) [9][10]. Among these, ED plays a pivotal role in the increase CV morbidity and mortality. The endothelium, previously considered a barrier selectively permeable between the blood and the vascular wall, is today recognized as a crucial organ in the regulation of the vascular tone and the structure [11]. Endothelial cells represent a complex and dynamic system able to respond to stimuli of different nature, having a large number and type of receptors and the capacity to produce a series of substances able to act on different pathways.
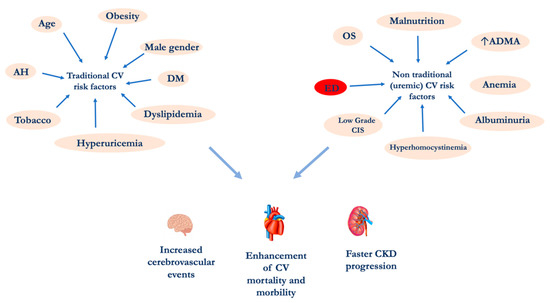
Figure 1. CV risk factors in CKD patients. Abbreviations: AH, Arterial Hypertension; ADMA Asymmetric dimethylarginine; CIS, chronic inflammatory state; CKD, chronic kidney disease; CV, cardiovascular; DM diabetes mellitus; ED, endothelial dysfunction; OS, oxidative stress.
Some authors have highlighted sexual dimorphism in endothelial cells [12][13]. These macro and micro vascular differences appear to be related to the protein synthesis by endothelial cells. In particular, the differences between the two sexes are: (i) the increased expression of androgen receptors, of estrogenic receptor (ER)-α and of vascular adhesion molecule-1 (VCAM-1) in men compared to women, (ii) the enhanced expression of platelet endothelial cell adhesion molecule-1 (PECAM-1), integrin αvβ3, intracellular adhesion molecule-1 (ICAM-1) and neural cadherin (N-CAD) in females, while ER-β and VE-cadherin (VE-CAD) do not show differences between the two sexes [14].
Physiological endothelial functions include the production of adhesion molecules, the platelet activation and the production of factors involved in the coagulation cascade and in the fibrinolytic system, the regulation of inflammatory response, the cell proliferation, and the control of the vascular tone. Endothelial cells constitute a real endocrine-autocrine-paracrine organ [15][16]. The endothelium, therefore, exerts a significant role in the process of angiogenesis, in the vessel permeability, in the regulation of platelet activation and in the inflammation [17][18].
Recent studies have underlined the role that physical exercise and natural bioactive compounds (NBCs) may have separately in the prevention of ED in CKD patients [19][20][21][22]. Nevertheless, nowadays there are few studies in the literature that investigate the effects induced by the physical exercise associated with NBCs assumption in CKD patients [23][24][25][26].
Some authors demonstrated how NBCs (such as curcumin) and the regular practice of aerobic exercise significantly improve endothelial function [23]. Akazawa et al. compared the assumption of 150 mg of curcumin per day for 8 weeks with the practice of aerobic exercise training more than 3 days per week for 8 weeks (2–3 supervised sessions and additional home-based training) on vascular endothelial function. The authors divided the study population in three subgroups: (i) curcumin group, (ii) exercise group and (iii) control group. Flow-mediated dilation increased significantly and equally in the curcumin and exercise groups, while no changes were detected in the control group. In this study, the authors evaluated separately the effects of the two interventions, but it may be interesting to understand how the combined effects could impact on ED.
2. Chronic Kidney Disease and Endothelial Dysfunction
Chronic nephropathy is a systemic disease that currently affects about 10% of the general population [27]. The CKD increased prevalence is certainly due to a global ageing of the population and especially to the increase in the incidence of metabolic diseases such as diabetes mellitus (DM), metabolic syndrome (MetS), obesity, etc. [28].
The association between CKD and CVD was first highlighted in end-stage renal diseases (ESRD) patients in dialytic treatment [2]. More recently, it has been shown that even the presence of slight changes in renal function, such as albuminuria, leads to a significant enhancement in CV events, which are more frequent compared to the progression versus ESRD.
CKD causes ED through several mechanisms. Physiologically, the vascular endothelium can be considered a “real organ”, able to secrete, in response to a great variety of signals, numerous chemical mediators [29][30]. Among the main physiological functions of the endothelium, the nitric oxide (NO) release represents a milestone. This molecule displays several functions in the human body such as the regulation of neuronal communication, inflammatory and immune responses, and the regulation of vascular tone [31][32].
ED represents an abnormality that develops at the level of the tunic that covers the internal surface of arterial and venous vessels and more precisely an alteration of normal endothelium, which results in the loss of some structural and/or functional characteristics [33][34].
In particular, at renal level it is possible to distinguish different types of endothelial cells, such as those of the small vessels that regulate the blood flow in the kidney and are also involved in the coagulation process and in the regulation of vascular permeability and in the inflammatory state. The glomerular endothelial cells, in addition to the already mentioned functions, are involved in glomerular filtration and in providing support to the podocytes. Finally, the endothelial cells of the microvasculature in the kidney tubules participate in the tubular reabsorption [35][36]. As for the kidney dysfunction, it has been highlighted a transition of kidney endothelial cells into mesenchymal phenotype, phenomenon called endothelial-to-mesenchymal transition, promoting renal fibrosis. Therefore, in CKD patients the accumulation of uremic toxins worsens the residual renal function and induces the systemic ED, contributing to cause CV comorbidities [37]. In fact, the increased incidence of CV events in CKD patients is related to the uremic toxins but also to the chronic inflammation state, typical of this pathological condition [38]. The evidence of a close relationship between kidney and hearth lays the foundations for a wider and more detailed stratification of the CV risk and suggests the opportunity for a correct and prompt management of CV comorbidities, starting from the early stages of CKD [39].
During CKD, the ED development appears to be related to a decrease in the inducible nitric oxide synthase (iNOS) activity and an increase in ADMA levels [31]. The latter gradually increases with the decline of the renal function. In fact, ADMA concentration is related to CKD progression, and it is an important CV risk factor both in the general population and in nephropathic patients [40]. ADMA is synthetized endogenously by the degradation of methylated proteins [41]. The asymmetrical residues of methylated arginine such as ADMA itself and NG-monomethyl-L-arginine (L-NMMA), inhibit the NO synthesis, as they are competitive inhibitors of NO synthase (NOS). ADMA and L-NMMA are excreted in the urine by renal pathways, and in CKD patients it can be observed an enhancement ADMA levels [42]. The ADMA accumulation in nephropathic patients could be related to renal parenchyma damage, resulting in a reduced expression and activity of dimethylarginine dimethylaminohydrolase (DDAH) [43]. This enzyme is involved in ADMA metabolism, in fact DDAH induces the ADMA hydrolytic degradation into L-citrulline and dimethylamine [44]. In some pathological conditions, such as DM, hyperhomocysteinemia, OS and dyslipidemia, DDAH reduced levels have been observed [45]. A decreased activity of DDAH induces an increase in ADMA concentration and subsequently a decreased NO production in the endothelium [46]. For this reason, ADMA has been described as a compound able to induce ED. In fact, intra-arterial local infusion of ADMA seems significantly reduce the blood flow in forearm [47], while the intravenous one causes an enhancement of blood pressure values of 6% and of systemic vascular resistance of 24% [48]. Another cause of ADMA accumulation in CKD patients, it is represented by an increased rate of protein turnover that, in turn, causes enhanced concentrations of methylated protein [49]. A study by Damaso et al. [50] conducted on a cohort of 225 HD patients, highlighted also an interaction between inflammation biomarkers and ADMA. In particular, the mortality was higher in the group that had both elevated ADMA and C-reactive protein (CRP) levels compared to groups that had only one of the biomarkers altered. The authors concluded that inflammation amplifies the risk of death and CV events in HD patients with ADMA high levels. Therefore, further studies are needed to establish normal serum and urinary ranges of ADMA for different ages. The link between ED, increased ADMA levels and OS is represented by the impaired NO production. In fact, NO, as above mentioned, performs several biological functions [51]. The reaction catalyzed by NOS enzyme, starting from L-arginine, produces NO and L-citrulline [52][53][54]. In blood vessels, endothelial NOS (eNOS) represent the most abundant isozyme. Under pathological conditions, eNOS can produce reactive oxygen species (ROS) [55]. In turn, the OS can induce an alteration in NO production through two mechanisms involving eNOS enzyme: the first one is represented by the eNOS inhibition and the second one is due to uncoupling eNOS. Consequently, the decreased NO production contributes to cause ED [56]. Among the factors related to OS during CKD, it should be considered the accumulation of uremic toxins such as indoxyl sulfate (IS), homocysteine (Hcy), and advanced glycation end products (AGEs) [57][58].
In nephropathic patients, another factor related to ED is hyperhomocysteinemia (Figure 2) [59]. Hcy is an amino acid sulfide, placed at the crossroad of a complex metabolic pathway. Normal Hcy levels are between 5 and 15 μmol/L [60], while hyperhomocysteinemia is defined when Hcy concentration is above 15 μmol/L [61].
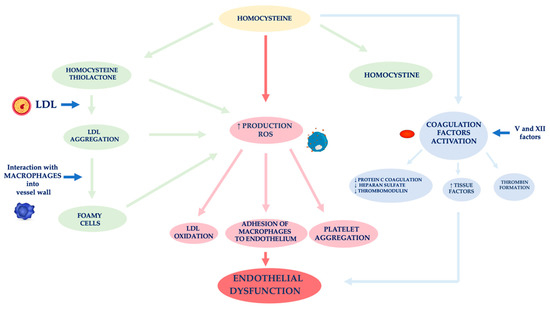
Figure 2. Mechanisms of vascular wall damage induced by homocysteine. Abbreviations: LDL, low-density lipoprotein; ROS, reactive oxygen species.
Factors affecting an increase of Hcy plasma levels can be genetic and/or acquired. As for the former, several mutations have been identified in the various genes involved in Hcy metabolism. The most analyzed mutation is certainly that relating to the methylenetetrahydrofolate reductase (MTHFR) gene. The acquired factors include gender, age, kidney function and lifestyle [62]. In fact, Hcy levels in CKD patients are on average increased because of its reduced kidney metabolism, namely between 20 and 40 μmol/L, especially if it is also present a folate and B12 vitamin deficiency, malnutrition, accumulation of uremic toxins [63][64][65][66]. Several authors demonstrated that Hcy plasma concentration is directly related to plasma creatinine values as it has been shown that in the renal parenchyma there are enzymes involved in Hcy remethylation and transsulfuration [67][68][69]. Jansen et al. hypothesized that hyperhomocyteinemia in CKD patients is mainly related to the slowing of its catabolic pathway of transsulfuration [70]. In fact, the enzymes cystathionine-β-synthase and cystathionine-γ-lyase are located in the proximal tubules of the nephron. However, the Hcy excretion in the urine is limited to 0.1% of the Hcy total, as the latter is largely reabsorbed at the tubular level. Unlike cystine, which has an antioxidant action, Hcy has a pro-oxidant action, as it promotes the formation of free radicals. The different behavior of these two amino acids is related to both an Hcy auto-oxidation and its enzymatic transformation into thiolactone, a substance with a strong oxidizing power [71]. The formation of thiolactone induces structural changes in intracellular proteins up to the loss of the biological activity of native proteins. The thiolation process can also occur at level of extracellular components, such as apolipoprotein B (apoB) and low-density lipoprotein (LDL). Hcy forms, with the LDL, aggregates which are captured by macrophages in the vessel wall. These phagocytic cells are transformed into foamy cells that release other free radicals that induce the oxidation of LDL, the platelet aggregation and the adhesion of macrophages to the endothelium [72][73][74]. Meanwhile, the Hcy autoxidation induces the production of further free radicals (hydroxyl) that trigger a process of lipid peroxidation at the level of the endothelial membranes [75][76].
ED induced by Hcy is also caused by the activation of coagulation factors, such as V and XII, whose stimulation induces: (i) a reduced expression of the coagulation protein C, of the thrombomodulin and of the heparan sulfate by the endothelium, (ii) an increased expression of the tissue factors and the thrombin formation [77]. In physiological conditions, the endothelium develops defensive mechanisms against the toxic action induced by hyperhomocysteinemia, releasing NO which forms S-nitrous-homocysteine, a compound that inhibits the production of hydrogen peroxide. S-nitrous-homocysteine has an important vasodilating action and inhibits platelet aggregation [78]. However, the persistence of the damage produced by a chronic condition of hyperhomocysteinemia, progressively reduces the ability of the endothelium to produce NO. An in vitro study by Tyagi et al. showed that Hcy activates the protease-activated receptors 4 which induces the production of ROS through an increase in NADPH oxidase and a decrease in expression of thioredoxin. Hyperhomocysteinemia also reduces the bioavailability of NO through the increase in NO2-tyrosine and through the accumulation of ADMA caused by the reduced expression of DDAH [79].
Furthermore, Hcy appears to stimulate the release of interleukin (IL)-8 and macrophage chemotactic factor (MCF). These chemokines have specific chemotactic activity for monocytes and neutrophils. The infiltration of the arterial wall by monocytes is a key event for the induction of atherogenesis. Instead, the monocyte chemoattractant protein (MCP) stimulates the migration of monocytes into the intima of the vessel wall. The OS induced by Hcy, on the one hand, directly damages endothelial cells, and on the other hand, induces the expression of matrix metalloproteases (MMPs), enzymes responsible for remodeling the vessel wall. In physiological conditions, MMPs are in equilibrium with their inhibitors, while in pathological conditions, this equilibrium is unbalanced towards a decrease in MMPs contextual to the increase in their inhibitors [80][81].
Another mechanism causing ED in CKD is an unbalance of the calcium-phosphorus metabolism. In particular, phosphate retention induces the development of CKD-mineral bone disorder (CKD-MBD) which, in turn, contributes to cause vascular calcifications. In fact, numerous epidemiological studies have shown that higher serum phosphorus levels, even in the absence of CKD, represent a risk factor for CVDs [82][83]. During CKD, vascular calcifications of the intima and media are frequent and are correlated to vascular rigidity. Elevated phosphate levels trigger the transformation of smooth muscle cells of the arterial wall into an osteoblast-like phenotype [84][85]. In addition, hyperphosphatemia affects ED, increasing apoptosis, inducing an increased production of ROS, impairing the NO production and decreasing the expression of annexin II [86][87]. The latter is a glycoprotein involved in various cellular functions, including the motility of epithelial cells, the fibrinolysis, the formation of anion channels and the interaction with matrix cells [88].
References
- Jacobs, C. Ethical problems posed by treatments of terminal chronic uremia. Presse Med. 1996, 25, 1359–1362.
- Cozzolino, M.; Mangano, M.; Stucchi, A.; Ciceri, P.; Conte, F.; Galassi, A. Cardiovascular disease in dialysis patients. Nephrol. Dial. Transplant. 2018, 33, iii28–iii34.
- Foley, R.N. Clinical epidemiology of cardiovascular disease in chronic kidney disease. J. Ren. Care 2010, 36, 4–8.
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522.
- Alani, H.; Tamimi, A.; Tamimi, N. Cardiovascular co-morbidity in chronic kidney disease: Current knowledge and future research needs. World J. Nephrol. 2014, 3, 156–168.
- Dessi, M.; Noce, A.; Dawood, K.F.; Galli, F.; Taccone-Gallucci, M.; Fabrini, R.; Bocedi, A.; Massoud, R.; Fucci, G.; Pastore, A.; et al. Erythrocyte glutathione transferase: A potential new biomarker in chronic kidney diseases which correlates with plasma homocysteine. Amino Acids 2012, 43, 347–354.
- Bocedi, A.; Noce, A.; Marrone, G.; Noce, G.; Cattani, G.; Gambardella, G.; Di Lauro, M.; Di Daniele, N.; Ricci, G. Glutathione Transferase P1-1 an Enzyme Useful in Biomedicine and as Biomarker in Clinical Practice and in Environmental Pollution. Nutrients 2019, 11, 1741.
- Roselli, M.; Guadagni, F.; Buonomo, O.; Belardi, A.; Ferroni, P.; Diodati, A.; Anselmi, D.; Cipriani, C.; Casciani, C.U.; Greiner, J.; et al. Tumor markers as targets for selective diagnostic and therapeutic procedures. Anticancer Res. 1996, 16, 2187–2192.
- Vallianou, N.G.; Mitesh, S.; Gkogkou, A.; Geladari, E. Chronic Kidney Disease and Cardiovascular Disease: Is there Any Relationship? Curr. Cardiol. Rev. 2019, 15, 55–63.
- Karpman, D.; Stahl, A.L.; Arvidsson, I. Extracellular vesicles in renal disease. Nat. Rev. Nephrol. 2017, 13, 545–562.
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069.
- Laughlin, M.H.; Welshons, W.V.; Sturek, M.; Rush, J.W.; Turk, J.R.; Taylor, J.A.; Judy, B.M.; Henderson, K.K.; Ganjam, V.K. Gender, exercise training, and eNOS expression in porcine skeletal muscle arteries. J. Appl. Physiol. 2003, 95, 250–264.
- Simmons, C.A.; Grant, G.R.; Manduchi, E.; Davies, P.F. Spatial heterogeneity of endothelial phenotypes correlates with side-specific vulnerability to calcification in normal porcine aortic valves. Circ. Res. 2005, 96, 792–799.
- Huxley, V.H.; Kemp, S.S.; Schramm, C.; Sieveking, S.; Bingaman, S.; Yu, Y.; Zaniletti, I.; Stockard, K.; Wang, J. Sex differences influencing micro- and macrovascular endothelial phenotype in vitro. J. Physiol. 2018, 596, 3929–3949.
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109.
- Ait-Oufella, H.; Maury, E.; Lehoux, S.; Guidet, B.; Offenstadt, G. The endothelium: Physiological functions and role in microcirculatory failure during severe sepsis. Intensive Care Med. 2010, 36, 1286–1298.
- Kruger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411.
- Schinzari, F.; Iantorno, M.; Campia, U.; Mores, N.; Rovella, V.; Tesauro, M.; Di Daniele, N.; Cardillo, C. Vasodilator responses and endothelin-dependent vasoconstriction in metabolically healthy obesity and the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E787–E792.
- Headley, S.; Germain, M.; Milch, C.; Pescatello, L.; Coughlin, M.A.; Nindl, B.C.; Cornelius, A.; Sullivan, S.; Gregory, S.; Wood, R. Exercise training improves HR responses and V O2peak in predialysis kidney patients. Med. Sci. Sports Exerc. 2012, 44, 2392–2399.
- Kirkman, D.L.; Ramick, M.G.; Muth, B.J.; Stock, J.M.; Pohlig, R.T.; Townsend, R.R.; Edwards, D.G. Effects of aerobic exercise on vascular function in nondialysis chronic kidney disease: A randomized controlled trial. Am. J. Physiol. Ren. Physiol. 2019, 316, F898–F905.
- Romani, A.; Bernini, R.; Noce, A.; Urciuoli, S.; Di Lauro, M.; Pietroboni Zaitseva, A.; Marrone, G.; Di Daniele, N. Potential Beneficial Effects of Extra Virgin Olive Oils Characterized by High Content in Minor Polar Compounds in Nephropathic Patients: A Pilot Study. Molecules 2020, 25, 4757.
- Noce, A.; Marrone, G.; Urciuoli, S.; Di Daniele, F.; Di Lauro, M.; Pietroboni Zaitseva, A.; Di Daniele, N.; Romani, A. Usefulness of Extra Virgin Olive Oil Minor Polar Compounds in the Management of Chronic Kidney Disease Patients. Nutrients 2021, 13, 581.
- Akazawa, N.; Choi, Y.; Miyaki, A.; Tanabe, Y.; Sugawara, J.; Ajisaka, R.; Maeda, S. Curcumin ingestion and exercise training improve vascular endothelial function in postmenopausal women. Nutr. Res. 2012, 32, 795–799.
- Ali, B.H.; Karaca, T.; Al Suleimani, Y.; Al Za’abi, M.; Al Kalbani, J.; Ashique, M.; Nemmar, A. The effect of swimming exercise on adenine-induced kidney disease in rats, and the influence of curcumin or lisinopril thereon. PLoS ONE 2017, 12, e0176316.
- Zanuzo, K.; Guareschi, Z.M.; Detogni, A.C.; Huning, L.P.; Rodrigues, P.F.; Porto, E.M.; Grassiolli, S.; Amorim, J.P.A. Physical exercise associated with vitamin D chronic supplementation reduces kidney injury induced by monosodium glutamate. An. Acad. Bras. Cienc. 2020, 92, e20201097.
- Kutlu, M.; Naziroglu, M.; Simsek, H.; Yilmaz, T.; Sahap Kukner, A. Moderate exercise combined with dietary vitamins C and E counteracts oxidative stress in the kidney and lens of streptozotocin-induced diabetic-rat. Int. J. Vitam. Nutr. Res. 2005, 75, 71–80.
- Thomas, R.; Kanso, A.; Sedor, J.R. Chronic kidney disease and its complications. Prim. Care 2008, 35, 329–344.
- Petrie, J.R.; Guzik, T.J.; Touyz, R.M. Diabetes, Hypertension, and Cardiovascular Disease: Clinical Insights and Vascular Mechanisms. Can. J. Cardiol. 2018, 34, 575–584.
- Tousoulis, D.; Kampoli, A.M.; Tentolouris, C.; Papageorgiou, N.; Stefanadis, C. The role of nitric oxide on endothelial function. Curr. Vasc. Pharmacol. 2012, 10, 4–18.
- Menghini, R.; Campia, U.; Tesauro, M.; Marino, A.; Rovella, V.; Rodia, G.; Schinzari, F.; Tolusso, B.; di Daniele, N.; Federici, M.; et al. Toll-like receptor 4 mediates endothelial cell activation through NF-kappaB but is not associated with endothelial dysfunction in patients with rheumatoid arthritis. PLoS ONE 2014, 9, e99053.
- Aldamiz-Echevarria, L.; Andrade, F. Asymmetric dimethylarginine, endothelial dysfunction and renal disease. Int. J. Mol. Sci. 2012, 13, 11288–11311.
- Nitsch, D.D.; Ghilardi, N.; Muhl, H.; Nitsch, C.; Brune, B.; Pfeilschifter, J. Apoptosis and expression of inducible nitric oxide synthase are mutually exclusive in renal mesangial cells. Am. J. Pathol. 1997, 150, 889–900.
- Jamwal, S.; Sharma, S. Vascular endothelium dysfunction: A conservative target in metabolic disorders. Inflamm. Res. 2018, 67, 391–405.
- Hadi, H.A.; Carr, C.S.; Al Suwaidi, J. Endothelial dysfunction: Cardiovascular risk factors, therapy, and outcome. Vasc. Health Risk Manag. 2005, 1, 183–198.
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 2019, 15, 87–108.
- Verma, S.K.; Molitoris, B.A. Renal endothelial injury and microvascular dysfunction in acute kidney injury. Semin. Nephrol. 2015, 35, 96–107.
- Jourde-Chiche, N.; Dou, L.; Cerini, C.; Dignat-George, F.; Brunet, P. Vascular incompetence in dialysis patients—Protein-bound uremic toxins and endothelial dysfunction. Semin. Dial. 2011, 24, 327–337.
- Podkowinska, A.; Formanowicz, D. Chronic Kidney Disease as Oxidative Stress- and Inflammatory-Mediated Cardiovascular Disease. Antioxidants 2020, 9, 752.
- Cobo, G.; Lindholm, B.; Stenvinkel, P. Chronic inflammation in end-stage renal disease and dialysis. Nephrol. Dial. Transplant. 2018, 33, iii35–iii40.
- Liu, X.; Xu, X.; Shang, R.; Chen, Y. Asymmetric dimethylarginine (ADMA) as an important risk factor for the increased cardiovascular diseases and heart failure in chronic kidney disease. Nitric Oxide 2018, 78, 113–120.
- McDermott, J.R. Studies on the catabolism of Ng-methylarginine, Ng, Ng-dimethylarginine and Ng, Ng-dimethylarginine in the rabbit. Biochem. J. 1976, 154, 179–184.
- Boger, R.H.; Zoccali, C. ADMA: A novel risk factor that explains excess cardiovascular event rate in patients with end-stage renal disease. Atheroscler. Suppl. 2003, 4, 23–28.
- Sitar, M.E. Asymmetric Dimethylarginine and Its Relation As a Biomarker in Nephrologic Diseases. Biomark. Insights 2016, 11, 131–137.
- Ogawa, T.; Kimoto, M.; Sasaoka, K. Occurrence of a new enzyme catalyzing the direct conversion of NG,NG-dimethyl-L-arginine to L-citrulline in rats. Biochem. Biophys. Res. Commun. 1987, 148, 671–677.
- Ito, A.; Tsao, P.S.; Adimoolam, S.; Kimoto, M.; Ogawa, T.; Cooke, J.P. Novel mechanism for endothelial dysfunction: Dysregulation of dimethylarginine dimethylaminohydrolase. Circulation 1999, 99, 3092–3095.
- Sibal, L.; Agarwal, S.C.; Home, P.D.; Boger, R.H. The Role of Asymmetric Dimethylarginine (ADMA) in Endothelial Dysfunction and Cardiovascular Disease. Curr. Cardiol. Rev. 2010, 6, 82–90.
- Calver, A.; Collier, J.; Leone, A.; Moncada, S.; Vallance, P. Effect of local intra-arterial asymmetric dimethylarginine (ADMA) on the forearm arteriolar bed of healthy volunteers. J. Hum. Hypertens. 1993, 7, 193–194.
- Achan, V.; Broadhead, M.; Malaki, M.; Whitley, G.; Leiper, J.; MacAllister, R.; Vallance, P. Asymmetric dimethylarginine causes hypertension and cardiac dysfunction in humans and is actively metabolized by dimethylarginine dimethylaminohydrolase. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1455–1459.
- Zoccali, C.; Kielstein, J.T. Asymmetric dimethylarginine: A new player in the pathogenesis of renal disease? Curr. Opin. Nephrol. Hypertens. 2006, 15, 314–320.
- Oliva-Damaso, E.; Oliva-Damaso, N.; Rodriguez-Esparragon, F.; Payan, J.; Baamonde-Laborda, E.; Gonzalez-Cabrera, F.; Santana-Estupinan, R.; Rodriguez-Perez, J.C. Asymmetric (ADMA) and Symmetric (SDMA) Dimethylarginines in Chronic Kidney Disease: A Clinical Approach. Int. J. Mol. Sci. 2019, 20, 3668.
- Gheibi, S.; Jeddi, S.; Kashfi, K.; Ghasemi, A. Regulation of vascular tone homeostasis by NO and H2S: Implications in hypertension. Biochem. Pharmacol. 2018, 149, 42–59.
- Su, Y. Regulation of endothelial nitric oxide synthase activity by protein-protein interaction. Curr. Pharm. Des. 2014, 20, 3514–3520.
- Korhonen, R.; Lahti, A.; Kankaanranta, H.; Moilanen, E. Nitric oxide production and signaling in inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 471–479.
- Andrew, P.J.; Mayer, B. Enzymatic function of nitric oxide synthases. Cardiovasc. Res. 1999, 43, 521–531.
- Forstermann, U.; Li, H. Therapeutic effect of enhancing endothelial nitric oxide synthase (eNOS) expression and preventing eNOS uncoupling. Br. J. Pharmacol. 2011, 164, 213–223.
- Li, H.; Horke, S.; Forstermann, U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 2014, 237, 208–219.
- Wojtaszek, E.; Oldakowska-Jedynak, U.; Kwiatkowska, M.; Glogowski, T.; Malyszko, J. Uremic Toxins, Oxidative Stress, Atherosclerosis in Chronic Kidney Disease, and Kidney Transplantation. Oxid. Med. Cell. Longev. 2021, 2021, 6651367.
- Noce, A.; Rovella, V.; Marrone, G.; Cattani, G.; Zingaretti, V.; Limongi, D.; D’Agostini, C.; Sorge, R.; Casasco, M.; Di Daniele, N.; et al. Hemodialysis biomarkers: Total advanced glycation end products (AGEs) against oxidized human serum albumin (HSAox). Acta Diabetol. 2019, 56, 1323–1331.
- Jakovljevic, B.; Gasic, B.; Kovacevic, P.; Rajkovaca, Z.; Kovacevic, T. Homocystein as a risk factor for developing complications in chronic renal failure. Mater. Sociomed. 2015, 27, 95–98.
- Azzini, E.; Ruggeri, S.; Polito, A. Homocysteine: Its Possible Emerging Role in At-Risk Population Groups. Int. J. Mol. Sci. 2020, 21, 1421.
- Ganguly, P.; Alam, S.F. Role of homocysteine in the development of cardiovascular disease. Nutr. J. 2015, 14, 6.
- Baszczuk, A.; Kopczynski, Z. Hyperhomocysteinemia in patients with cardiovascular disease. Postepy Hig. Med. Dosw. (Online) 2014, 68, 579–589.
- Di Daniele, N.; Di Renzo, L.; Noce, A.; Iacopino, L.; Ferraro, P.M.; Rizzo, M.; Sarlo, F.; Domino, E.; De Lorenzo, A. Effects of Italian Mediterranean organic diet vs. low-protein diet in nephropathic patients according to MTHFR genotypes. J. Nephrol. 2014, 27, 529–536.
- Dessi, M.; Di Giovamberardino, G.; Pieri, M.; Noce, A.; Zenobi, R.; Di Daniele, N.; Pastore, A. Influence of dialysis techniques and alternate vitamin supplementation on homocysteine levels in patients with known MTHFR genotypes. Clin. Exp. Nephrol. 2015, 19, 140–145.
- Pastore, A.; Noce, A.; Di Giovamberardino, G.; De Stefano, A.; Calla, C.; Zenobi, R.; Dessi, M.; Di Daniele, N. Homocysteine, cysteine, folate and vitamin B(1)(2) status in type 2 diabetic patients with chronic kidney disease. J. Nephrol. 2015, 28, 571–576.
- Long, Y.; Nie, J. Homocysteine in Renal Injury. Kidney Dis. 2016, 2, 80–87.
- Satta, E.; Perna, A.F.; Lombardi, C.; Acanfora, F.; Violetti, E.; Romano, M.M.; Capasso, R.; Pisano, M.; Paduano, F.; De Santo, N.G. Hyperhomocysteinemia in chronic renal failure. G. Ital. Nefrol. 2006, 23, 480–489.
- Wollesen, F.; Brattstrom, L.; Refsum, H.; Ueland, P.M.; Berglund, L.; Berne, C. Plasma total homocysteine and cysteine in relation to glomerular filtration rate in diabetes mellitus. Kidney Int. 1999, 55, 1028–1035.
- Friedman, A.N.; Bostom, A.G.; Selhub, J.; Levey, A.S.; Rosenberg, I.H. The kidney and homocysteine metabolism. J. Am. Soc. Nephrol. 2001, 12, 2181–2189.
- Jansen, M.A.; Hart, A.A.; Korevaar, J.C.; Dekker, F.W.; Boeschoten, E.W.; Krediet, R.T.; Group, N.S. Predictors of the rate of decline of residual renal function in incident dialysis patients. Kidney Int. 2002, 62, 1046–1053.
- Zivkovic, V.; Jakovljevic, V.; Pechanova, O.; Srejovic, I.; Joksimovic, J.; Selakovic, D.; Barudzic, N.; Djuric, D.M. Effects of DL-homocysteine thiolactone on cardiac contractility, coronary flow, and oxidative stress markers in the isolated rat heart: The role of different gasotransmitters. Biomed. Res. Int. 2013, 2013, 318471.
- Jakubowski, H.; Zhang, L.; Bardeguez, A.; Aviv, A. Homocysteine thiolactone and protein homocysteinylation in human endothelial cells: Implications for atherosclerosis. Circ. Res. 2000, 87, 45–51.
- Gurda, D.; Handschuh, L.; Kotkowiak, W.; Jakubowski, H. Homocysteine thiolactone and N-homocysteinylated protein induce pro-atherogenic changes in gene expression in human vascular endothelial cells. Amino Acids 2015, 47, 1319–1339.
- Griffiths, H.R.; Aldred, S.; Dale, C.; Nakano, E.; Kitas, G.D.; Grant, M.G.; Nugent, D.; Taiwo, F.A.; Li, L.; Powers, H.J. Homocysteine from endothelial cells promotes LDL nitration and scavenger receptor uptake. Free Radic. Biol. Med. 2006, 40, 488–500.
- Nakano, E.; Taiwo, F.A.; Nugent, D.; Griffiths, H.R.; Aldred, S.; Paisi, M.; Kwok, M.; Bhatt, P.; Hill, M.H.; Moat, S.; et al. Downstream effects on human low density lipoprotein of homocysteine exported from endothelial cells in an in vitro system. J. Lipid Res. 2005, 46, 484–493.
- Bayes, B.; Pastor, M.C.; Bonal, J.; Junca, J.; Romero, R. Homocysteine and lipid peroxidation in haemodialysis: Role of folinic acid and vitamin E. Nephrol. Dial. Transplant. 2001, 16, 2172–2175.
- Tinelli, C.; Di Pino, A.; Ficulle, E.; Marcelli, S.; Feligioni, M. Hyperhomocysteinemia as a Risk Factor and Potential Nutraceutical Target for Certain Pathologies. Front. Nutr. 2019, 6, 49.
- Knipp, M.; Braun, O.; Vasak, M. Searching for DDAH inhibitors: S-nitroso-L-homocysteine is a chemical lead. J. Am. Chem. Soc. 2005, 127, 2372–2373.
- Tyagi, N.; Sedoris, K.C.; Steed, M.; Ovechkin, A.V.; Moshal, K.S.; Tyagi, S.C. Mechanisms of homocysteine-induced oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2649–H2656.
- Raffetto, J.D.; Khalil, R.A. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem. Pharmacol. 2008, 75, 346–359.
- Wang, X.; Khalil, R.A. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv. Pharmacol. 2018, 81, 241–330.
- Razzaque, M.S. Phosphate toxicity: New insights into an old problem. Clin. Sci. 2011, 120, 91–97.
- Noce, A.; Canale, M.P.; Capria, A.; Rovella, V.; Tesauro, M.; Splendiani, G.; Annicchiarico-Petruzzelli, M.; Manzuoli, M.; Simonetti, G.; Di Daniele, N. Coronary artery calcifications predict long term cardiovascular events in non diabetic Caucasian hemodialysis patients. Aging 2015, 7, 269–279.
- Giachelli, C.M. Vascular calcification mechanisms. J. Am. Soc. Nephrol. 2004, 15, 2959–2964.
- Felsenfeld, A.J.; Levine, B.S.; Rodriguez, M. Pathophysiology of Calcium, Phosphorus, and Magnesium Dysregulation in Chronic Kidney Disease. Semin. Dial. 2015, 28, 564–577.
- Di Marco, G.S.; Konig, M.; Stock, C.; Wiesinger, A.; Hillebrand, U.; Reiermann, S.; Reuter, S.; Amler, S.; Kohler, G.; Buck, F.; et al. High phosphate directly affects endothelial function by downregulating annexin II. Kidney Int. 2013, 83, 213–222.
- Burger, D.; Levin, A. ‘Shedding’ light on mechanisms of hyperphosphatemic vascular dysfunction. Kidney Int. 2013, 83, 187–189.
- Fang, Y.T.; Lin, C.F.; Liao, P.C.; Kuo, Y.M.; Wang, S.; Yeh, T.M.; Shieh, C.C.; Su, I.J.; Lei, H.Y.; Lin, Y.S. Annexin A2 on lung epithelial cell surface is recognized by severe acute respiratory syndrome-associated coronavirus spike domain 2 antibodies. Mol. Immunol. 2010, 47, 1000–1009.
More
Information
Subjects:
Food Science & Technology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
885
Revisions:
2 times
(View History)
Update Date:
25 Aug 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No