Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Frank Aboubakar Nana | + 2985 word(s) | 2985 | 2021-08-13 06:05:19 | | | |
| 2 | Vivi Li | Meta information modification | 2985 | 2021-08-19 03:36:28 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Aboubakar Nana, F. Focal Adhesion Kinase. Encyclopedia. Available online: https://encyclopedia.pub/entry/13321 (accessed on 07 February 2026).
Aboubakar Nana F. Focal Adhesion Kinase. Encyclopedia. Available at: https://encyclopedia.pub/entry/13321. Accessed February 07, 2026.
Aboubakar Nana, Frank. "Focal Adhesion Kinase" Encyclopedia, https://encyclopedia.pub/entry/13321 (accessed February 07, 2026).
Aboubakar Nana, F. (2021, August 18). Focal Adhesion Kinase. In Encyclopedia. https://encyclopedia.pub/entry/13321
Aboubakar Nana, Frank. "Focal Adhesion Kinase." Encyclopedia. Web. 18 August, 2021.
Copy Citation
Small-cell lung cancer (SCLC) represents 15% of all lung cancers and it is clinically the most aggressive type, being characterized by a tendency for early metastasis, with two-thirds of the patients diagnosed with an extensive stage (ES) disease and a five-year overall survival (OS) as low as 5%. There are still no effective targeted therapies in SCLC despite improved understanding of the molecular steps leading to SCLC development and progression these last years.
focal adhesion kinase
small-cell lung cancer
targeted therapy
1. Introduction
Lung cancer, which arises from lung epithelial cells, is histologically divided into two main types: small-cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC), which represent 15% and 85% of the cases, respectively [1]. As opposed to SCLC, oncogenic drivers with sensitivity to targeted therapies have been discovered in NSCLC. Tyrosine kinase inhibitors (TKIs) targeting epidermal growth factor receptor (EGFR) mutations, anaplastic lymphoma kinase (ALK) rearrangements, or other oncogenic abnormalities have brought remarkable improvements in the outcome of oncogenic-driven NSCLC patients [2]. Immunotherapy with anti-programmed death-(ligand) 1 (PD-(L)1) immune checkpoint inhibitors (ICIs) has also significantly improved the survival of NSCLC patients without oncogenic drivers [3][4][5][6][7][8][9]. Clinically, SCLC is the most aggressive type of lung cancer, being characterized by a high growth rate, a fast doubling time, and a tendency for early metastasis, with two-thirds of the patients diagnosed with an extensive stage (ES) disease [10][11]. While a good initial response to chemotherapy and/or radiation therapy is observed in most patients, they typically recur or progress rapidly after the primary treatment, with a median overall survival (OS) of 24–38 months in limited stage (LS) [12][13] and 7–10 months in ES [14], and a five-year OS as low as 5% [1].
Despite improvements in the understanding of the molecular steps that lead to SCLC development and progression these last years, there are still no effective targeted therapies in SCLC. Rovalpituzumab tesirine (Rova-T) is an antibody-drug conjugate (pyrrolobenzodiazepine (PBD)-dimer cytotoxic) that is directed against Delta-like 3 (DLL3), an inhibitory NOTCH ligand, which has been shown to be overexpressed on the surface of SCLC cells [15]. Despite encouraging preclinical and early clinical results, targeted therapy with Rova-T underperformed in the phase II TRINITY trial, including pretreated SCLC patients with high levels of DLL3 on tumor cell surface [15][16]. After four decades, the only modest improvement in the OS of patients suffering from ES-SCLC has recently been shown in a trial combining atezolizumab, an anti-PD-L1 ICI, with carboplatin and etoposide, chemotherapy agents [17]. In this trial, the OS was 10.3 months in the chemotherapy alone arm, while it was 12.3 months in the chemotherapy plus immunotherapy arm. Based on this positive trial, atezolizumab that is associated to carboplatin an etoposide recently became the new standard of care in the first-line treatment of ES-SCLC [17]. At relapse or progression after a first-line treatment, a rechallenge with platinum and etoposide is proposed to tumors that are considered to be sensitive to platinum (relapse or progression within 60 or 90 days of completion of chemotherapy) [18], while a second-line chemotherapy with topotecan is proposed to tumors platinum-refractory (relapse or progression before three to six months). However, the response rates are poor and OS ranges from 1.2 months to 7.6 months based on systematic reviews of real-world data 15 [19]. These disappointing results highlight the need for novel therapies.
Focal adhesion kinase (FAK) is a 125 kDa non-receptor protein tyrosine kinase that is known to be overexpressed and activated in several cancers, including SCLC [20][21][22][23][24][25][26][27][28]. Unlike receptor tyrosine kinases (RTKs), such as epidermal growth factor receptor (EGFR), non-RTKs, such as FAK, are cytoplasmic enzymes that lack transmembrane and extracellular domains [29]. FAK localizes to focal adhesions and it is triggered off by extracellular signals, such as integrin-mediated adhesion and some growth factors [30]. Therefore, FAK plays a central role in the interaction between cells, including cancer cells and their microenvironment. The FAK structure includes an NH2-terminal Protein4.1-ezrin-radixin-moesin (FERM) domain, a central kinase domain, two proline-rich motifs, and a COOH-terminal focal adhesion targeting (FAT) domain. FAK is maintained in an inactive state by the binding of the FERM domain to the kinase domain, which blocks access to the catalytic site and sequesters the activation loop, as well as the key autophosphorylation site tyrosine 397 (Tyr397) (Figure 1). The engagement of integrins with the extracellular matrix (ECM) or growth factors leads to signals that displace the FERM domain, resulting in rapid autophosphorylation of Tyr397, which is a critical event in signal transduction by FAK [30][31]. Tyr397 phosphorylation provides a binding site that recruits and activates Src through the SH2 domains of Src family kinases. The FAK-Src complex therefore maintains Src and FAK in their activated states, creating a functional kinase complex [32].
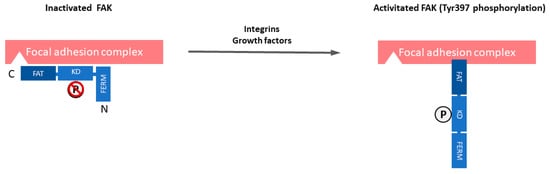
Figure 1. The domain organization and activation of focal adhesion kinase (FAK). FAK is composed of a central kinase domain (KD), an amino-terminal side that is flanked by a protein band 4.1-ezrin-radixin-moesin (FERM) homology domain, and a carboxy-terminal focal adhesion targeting (FAT) domain flanked by proline-rich regions (PRRs). FAK localizes to focal adhesions and is triggered off by extracellular signals such as integrin-mediated adhesion and some growth factors. FAK is maintained in an inactive state by the binding of the FERM domain to the kinase domain, which blocks access to the catalytic site and sequesters the activation loop, as well as the key autophosphorylation site tyrosine 397 (Tyr397). Engagement of integrins with the extracellular matrix (ECM) or growth factors leads to signals that displace the FERM domain, resulting in rapid autophosphorylation of Tyr397, which is a critical event in signal transduction by FAK.
Based on FAK overexpression and/or increased activity in cancer and its known function in multiple biological processes that play a role in the development and progression of cancers, such as crosstalk between cell and his microenvironment, cell growth, survival, adhesion, spreading, migration, invasion, angiogenesis, DNA damage repair, radioresistance, and regulation of cancer stem cells, it has been suggested that increased the expression and/or activity of FAK may have a critical role in cancer development and progression [33]. Therefore, FAK is a potential target for anti-cancer therapy, especially in SCLC, being known to be a highly invasive cancer. Small-molecule inhibitors targeting the FAK kinase domain and preventing FAK activation (Tyr397 autophosphorylation) have been developed. Phase I trials with GSK2256098 [34][35][36], VS-6062 [37], defactinib (VS-6063) [38][39][40], or BI853520 [41][42][43] have shown an acceptable safety profile and favorable pharmacokinetics. Most frequent treatment-related adverse events included digestive disorders (nausea, diarrhea, vomiting), headaches, reversible proteinuria, and unconjugated hyperbilirubinemia [34][35][36][37][38][39][40][41][42]. With GSK2256098, the best response of stable disease was observed in 37% of glioblastoma (three patients, median PFS 5, seven weeks) [36] and in 45% of advanced solid cancers (28 patients) [35]. With VS-6062, 34% of patients (31 patients) with advanced solid tumors exhibited stable disease at six weeks, including one case of SCLC for ≥6 cycles cycles [37]. VS-6063 led to the stabilization of advanced solid tumors in 43% of Caucasian patients (six cases) after six weeks of treatment [38] and in 33% of Asian patients (three cases) during more than 24 weeks (median PFS of 63 days) [40]. Recently, the combination of the FAK inhibitor GSK2256098 and the MEK inhibitor trametinib in recurrent advanced pancreatic ductal adenocarcinoma did not provide significant clinical activity in a phase II trial (PFS of 1.6 month and OS of 3.6 months) [44]. In malignant pleural mesothelioma, defactinib in maintenance after first-line chemotherapy in a phase II trial did not provide any benefit either (PFS of 4.1 months with defactinib vs 4.0 months with placebo, and OS of 12.7 months with defactinib vs. 13.6 months with placebo) [45]. Preoperative administration of defactinib in the ongoing phase II clinical trial NCT02004028 appears promising, with therapeutic activity (13% objective partial response, 67% stable disease, 17% tumor progression) and beneficial modification of the tumoral microenvironment [46]. Several clinical trials with defactinib associated with immunotherapy (NCT02758587, NCT03727880, NCT02943317), RAK/MEK inhibitor (NCT03875820), or chemotherapy (NCT02546531) are ongoing, with some of them being open to SCLC inclusion (Table 1) [34][35][36][37][39][40][41][42][43][44][45][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61]. Other small-molecules targeting the protein-protein interactions between FAK and other proteins, such as VEGFR-3, called scaffolding inhibitors, have been developed and shown to induce antitumoral effects in preclinical studies. Further research is needed to find predictive biomarkers of response to FAK TKI alone or, probably more promising, in association with another drug.
Table 1. FAK inhibitors with anti-tumor activity in preclinical studies and clinical trials.
| Name | Type | Specificity | Cancers Targeted | Study Phase | References |
|---|---|---|---|---|---|
| TAE-226 Novartis | Kinase inhibitor ATP competitive | FAK, IGF-IR, c-Met, Pyk2 | Glioma, ovarian | Preclinical | [47][62] |
| PF-573,228 Pfizer | Kinase inhibitor ATP competitive | FAK | Prostate, breast | Preclinical | [48] |
| GSK2256098 GlaxoSmithKline | Kinase inhibitor ATP competitive Reversible | FAK, UGT1A1 | Solid tumors (ovarian, pancreatic, meningioma, glioblastoma, malignant pleural mesothelioma) | Clinical: phase I & II | [34][35][36][44][49] NCT00996671, NCT02523014 |
| NVP-TAC544 | Kinase inhibitor ATP competitive | FAK | N/A | Preclinical | [50] |
| VS-4718 (PND-1186) Verastem | Kinase inhibitor ATP competitive Reversible | FAK, Pyk2 | Solid tumors (pancreas, breast, ovarian), acute myeloid leukemia, B-cell acute lymphoblastic leukemia | Clinical: phase I | [51] |
| VS-6062 (PF-562271 and PF271) Verastem | Kinase inhibitor ATP competitive Reversible | FAK, CDK2/CyclinE, CDK3/CyclinE, CDK1/CyclinB, Pyk2 | Prostate, pancreatic, head and neck | Clinical: phase I | [37][52] |
| VS-6063 (Defactinib) Verastem | Kinase inhibitor ATP competitive | FAK, Pyk2 | NSCLC, pancreatic cancer, ovarian, malignant pleural mesothelioma, hematologic | Clinical: phase I/Ib & II | [38][39][40][45][53] NCT02758587 NCT02004028 NCT03875820 NCT03727880, NCT02943317, NCT02913716, NCT02465060, NCT02546531 |
| 1H-Pyrrolo(2,3-b) Merk Serono | Kinase inhibitor non-ATP competitive | Hinge region of FAK | N/A | Preclinical | [54] |
| C4 CureFAKtor Pharmaceuticals | Scaffold inhibitor | FAK /VEGFR-3 | Neuroblastoma, pancreatic, breast | Preclinical | [55][56][57] |
| Compound R2 (Roslins) CureFAKtor Pharmaceuticals | Scaffold inhibitor | FAK, p53 | Colon, reast | Preclinical | [58] |
| Y11 CureFAKtor Pharmaceuticals | Scaffold inhibitor | FAK Y397 site | Colon, breast | Preclinical | [59] |
| BI853520 | ATP competitive inhibitor | FAK | Malignant pleural mesothelioma, non-hematologic malignancies | Preclinical, clinical | [42][43][60] |
Abbreviations: CDK: Cyclin-dependent kinases 1, 2, 3; FAK: focal adhesion kinase; IGF-IR: insulin-like growth factor 1 (IGF-1) receptor; N/A: data not available; Pyk2: proline-rich tyrosine kinase 2; UGT1A1: UDP-glucuronosyltransferase 1-1; VEGFR-3: vascular endothelial growth factor receptor 3.
2. FAK Role in Proliferation, Cell Cycle, and Survival
FAK activation during cell adhesion protects cells from anoikis, a form of apoptosis that is induced by cell detachment from ECM, favouring cancer growth and metastasis [63]. FAK is implicated in several pathways that contribute to cell survival. Phosphorylated FAK at Tyr397 can bind PIK3R2, which leads to the activation of AKT that inhibits apoptosis by regulating various molecules. Among other mechanisms, there is the suppression of apoptosis by FAK through c-JUN kinase activation downstream of CAS [33] and the inhibition of RIP interaction with the death receptor complex [64].
FAK also induces cell proliferation through the stimulation of cell cycle progression. One of the mechanisms is the formation of FAK/Src complex that allows for Src to phosphorylate FAK at Tyr925 and mediate its interaction with Grb2, which leads to the activation of the RAS-MAPK signaling pathway [40]. Another mechanism involves the FAK-induced increased expression of cyclin D1 and decreased expression of cycline-dependent kinase (Cdk) inhibitor p21 [65][66][67][68]. Other cell cycle regulators, such as cyclin E, Cdk inhibitor p27, and Skp2, also mediate FAK regulation of cell cycle progression [69][70][71][72]. Moreover, FAK is important for tumor cell-induced remodeling of the tumor matrix, which produces a rigid microenvironment and facilitates cell proliferation [73].
Specifically, in SCLC cell lines, it has been shown that the inhibition of FAK activity with PF-573,228, a FAK TKI, decreased proliferation, DNA synthesis, induced cell-cycle arrest in G2-M phases, and increased apoptosis in the NCI-H82, NCI-H146, NCI-H196, and NCI-H446 SCLC cell lines [74]. Treatment with increasing concentrations of PF-228 (0.1 to 10 µM) dose-dependently decreased the FAK phosphorylation (Tyr397) in these four cell lines, without modifying total FAK expression, and the inhibition of FAK activity with 1 to 10 µM PF-228 significantly decreased their proliferation, also dose-dependently (p < 0.001 for all tested concentrations beside 1 µM in NCI-H196), as assessed by a WST-1 assay. Cell cycle analysis showed that PF-228 inhibited progression through cell cycle by significantly reducing the S phase and inducing cell cycle arrest in the G2/M phases in the four cell lines after 24h-treatment, dose-dependently (p < 0.001). PF-228 at concentrations of 1 to 5 µM also significantly induced apoptosis in the four cell lines, as demonstrated by a dose-dependent increase of PARP p85 expression by WB after 48h-treatment. This was confirmed by flow cytometry in NCI-H82 and NCI-H446 cell lines, with a significant increase of BrdU-positive and activated Caspase 3-positive cells after 48h-treatment (p < 0.001 for all tested concentrations, except 1 µM in NCI-H446 in the Caspase-3 assay). Genetic inhibition of FAK through stable transduction with FAK shRNA and/or FAK-related non-kinase (FRNK), a splice variant lacking the N-terminal and kinase domains of FAK, revealed that the FAK-targeting (FAT) domain (attached to focal adhesion complex, where it inhibits pro-proliferative proteins) was necessary to inhibit proliferation, cell cycle progression, and survival [74]. Indeed, FAK shRNA transduction did not affect these functions, while the restoration of FAT domain by FRNK transduction inhibited proliferation, DNA synthesis, and induced apoptosis in the evaluated SCLC cell lines. Additionally, while FAK shRNA transduction increased the active Rac1 level, FRNK re-expression in cells that were previously transduced with FAK shRNA decreased it. Therefore, this study not only suggested that FAK is important in SCLC biology, but also that targeting its kinase domain might have a therapeutic potential, while targeting its FAT domain might have Rac1-mediated pro-tumoral activity and thus should be avoided.
3. FAK Role in Adhesion, Migration, and Invasion
FAK induces morphological changes in cells, including the formation of podosomes or invadopodia, contributing to cell migration [75][76][77]. Moreover, cancer cells overexpressing FAK are able to invade tissues [78]. FAK overexpression contributes to the metastatic phenotype of cancer cells by promoting cell migration and invasion.
Cell migration is a complex process that consists of several coordinated events, including protrusion of the leading edge, adhesion of the leading edge to the substrate [79], translocation of the cell body, and release of the trailing edge [80]. Thus, a strict regulation of tension at specific times and in specific areas of the cell is required for cell migration [81][82], where FAK plays an important role by sensing the mechanical forces that are generated in or exerted on cells [83], and modulating cell responses to environmental stimuli. Once activated by integrins, G protein-coupled receptors ligands, or growth factors, FAK is autophosphorylated at Tyr397 and activates proteins, such as Src, p130CAS, paxillin, and PIK3R2 [84], to regulate adhesion turnover at the cell front, a process that is central to migration [84][85][86][87][88]. FAK is indeed required for the organization of the leading edge in migrating cells [89]. The formation of a complex between FAK and Src, leading to the phosphorylation of the adaptor molecule CAS by the FAK/Src complex [90][91][92][93][94], is one of the best characterized downstream signaling pathways that mediate FAK-stimulated cell migration. A second mechanism involves FAK interaction with PIK3 and an adaptor molecule, Grb7 [95][96]. A third mechanism involves the modulation of the assembly and disassembly of actin cytoskeleton through the effect of FAK on the Rho family GTPases. Among the Rho family GTPases, FAK/Src signaling has, in particular, been implicated in regulating the activities of Rac1 and RhoA.
Besides its role in cell migration, FAK promotes invasion in normal and cancer cells by various mechanisms. In one of them, FAK promotes the formation of the Src-CAS-Crk-Dock180 complex, which activates Rac1 and JNK, and leads to increased expression or activity of metalloproteinases 2 (MMP2) and 9 (MMP9) [75]. MMPs are concentrated and activated at actin-rich cell/ECM contacts, termed podosomes or invadopodia, which are distinct from focal adhesion. In another mechanism, FAK cooperates with Src to disrupt the E-cadherin-based intercellular adherens junctions [97], contributing to EMT and, therefore, to the invasive phenotype of metastatic carcinomas through increased cell migration and remodelling of the ECM microenvironment [98][99][100]. In SCLC cell lines, the pharmacologic inhibition of FAK with PF-573,228 decreased cell adhesion [28], as well as migration and invasion [74]. In NCI-H69, NCI-H146, and NCI-H209 SCLC cell lines, PF-573,228 induced a dose-dependent decrease of cell adhesion on laminin, with the effect becoming statistically significant at the concentration of 10 µM (NCI-H69: p = 3 × 10−4, NCI-H146, and NCI-H209: p = 1 × 10−4 as compared to DMSO) [28]. Moreover, a wound healing assay combined with time-lapse microscopy showed that PF-573,228 decreased the migration velocity of two SCLC cell lines with an adherent component, from 5 to 2.5 µm/min. in NCI-H196 (p = 0.0561) and from 9 to 4 µm/min. in NCI-H446 (p = 0.0916)) [75]. Modified Boyden chambers showed that PF-573,228, at a concentration of 3 µM, also inhibited invasion, with the number of invasive cells being able to migrate to the lower side of the insert separating the two Boyden chambers, decreasing from 150 to 50 per field (20× magnification) for NCI-H196 and from 45 to five per field for NCI-H446 [75].
4. FAK in Epithelial to Mesenchymal Transition
Through epithelial-to-mesenchymal transition (EMT), cancer cells acquire a more motile phenotype, promoting invasion, metastasis, but also conferring resistance to chemotherapies and targeted therapies. Epithelial cancers undergoing EMT acquire transient mesenchymal features, like Vimentin and N-cadherin, which are associated with the loss of epithelial markers E-cadherin and β-catenin [101]. EMT is correlated with poor outcomes in SCLC [102], such as in many other cancers. Identified mechanisms inducing EMT in SCLC include inactive Notch signaling [103], activated MET receptor signaling through hepatocyte growth factor [102], and activated TGFβ/Akt signaling [104].
While FAK-mediated EMT has not yet been explored in SCLC, its important role has been demonstrated in other cancers and non-malignant cells [105][106][107][108]. Impaired FAK functions lead to a defective mesenchymal phenotype during EMT. Hence, upon TGF β-induced EMT, hepatocyte cell lines transduced with FRNK, a genomic method for inhibiting FAK, underwent an incomplete mesenchymal transition, exhibiting a lack of mesenchymal markers MMP9 and fibronectin and a persistence of membrane-bound E-cadherin [105]. Mammary tumor cells with deficient FAK scaffolding function due to Pro 878/881 mutation also displayed incomplete mesenchymal phenotype with increased E-cadherin and decreased N-cadherin, Vimentin, and fibronectin in a mice model [106]. It was associated with decreased metastasis potential and decreased expression of EMT-inducing gene Snail 1 [106]. A similar reduction of Snail 1 in embryonic FAK-null cells has been associated with the inability to display mesenchymal cell characteristics, while the reexpression of FAK restored mesenchymal phenotype and Snail 1 level through PI3K/Akt signaling [107]. In ovarian cancer, FAK controls EMT by upregulating transcription factor KLF8 via the PI3K/Akt pathway [108]. It has been shown that transcription factors Snail 1 and KLF8 repress E-cadherin expression, promoting EMT in various normal and malignant cells [109][110][111]. The inhibition of FAK by a genetic or a pharmacologic method decreased the EMT features and aggressiveness in colorectal carcinoma cell lines [112][113] and triple negative breast cancer cell lines in vitro [114], but not in NSCLC cell lines in vitro [115].
References
- Govindan, R.; Page, N.; Morgensztern, D.; Read, W.; Tierney, R.; Vlahiotis, A.; Spitznagel, E.L.; Piccirillo, J. Changing Epidemiology of Small-Cell Lung Cancer in the United States Over the Last 30 Years: Analysis of the Surveillance, Epidemiologic, and End Results Database. J. Clin. Oncol. 2006, 24, 4539–4544.
- Recondo, G.; Facchinetti, F.; Olaussen, K.A.; Besse, B.; Friboulet, L. Making the first move in EGFR-driven or ALK-driven NSCLC: First-generation or next-generation TKI? Nat. Rev. Clin. Oncol. 2018, 15, 694–708.
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639.
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crino, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135.
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028.
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833.
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet Lond. Engl. 2017, 389, 255–265.
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092.
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051.
- Wang, J.C.; Sone, S.; Feng, L.; Yang, Z.G.; Takashima, S.; Maruyama, Y.; Hasegawa, M.; Kawakami, S.; Honda, T.; Yamanda, T. Rapidly growing small peripheral lung cancers detected by screening CT: Correlation between radiological appearance and pathological features. Br. J. Radiol. 2000, 73, 930–937.
- Thomas, A.; Pattanayak, P.; Szabo, E.; Pinsky, P. Characteristics and Outcomes of Small Cell Lung Cancer Detected by CT Screening. Chest 2018, 154, 1284–1290.
- Turrisi, A.T.; Kim, K.; Blum, R.; Sause, W.T.; Livingston, R.B.; Komaki, R.; Wagner, H.; Aisner, S.; Johnson, D.H. Twice-daily compared with once-daily thoracic radiotherapy in limited small-cell lung cancer treated concurrently with cisplatin and etoposide. N. Engl. J. Med. 1999, 340, 265–271.
- Kubota, K.; Hida, T.; Ishikura, S.; Mizusawa, J.; Nishio, M.; Kawahara, M.; Yokoyama, A.; Imamura, F.; Takeda, K.; Negoro, S.; et al. Etoposide and cisplatin versus irinotecan and cisplatin in patients with limited-stage small-cell lung cancer treated with etoposide and cisplatin plus concurrent accelerated hyperfractionated thoracic radiotherapy (JCOG0202): A randomised phase 3 study. Lancet Oncol. 2014, 15, 106–113.
- Pujol, J.L.; Daures, J.P.; Riviere, A.; Quoix, E.; Westeel, V.; Quantin, X.; Breton, J.L.; Lemarie, E.; Poudenx, M.; Milleron, B.; et al. Etoposide plus cisplatin with or without the combination of 4’-epidoxorubicin plus cyclophosphamide in treatment of extensive small-cell lung cancer: A French Federation of Cancer Institutes multicenter phase III randomized study. J. Natl. Cancer Inst. 2001, 93, 300–308.
- Saunders, L.R.; Bankovich, A.J.; Anderson, W.C.; Aujay, M.A.; Bheddah, S.; Black, K.; Desai, R.; Escarpe, P.A.; Hampl, J.; Laysang, A.; et al. A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci. Transl. Med. 2015, 7, 302ra136.
- Rudin, C.M.; Pietanza, M.C.; Bauer, T.M.; Ready, N.; Morgensztern, D.; Glisson, B.S.; Byers, L.A.; Johnson, M.L.; Burris, H.A.; Robert, F.; et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: A first-in-human, first-in-class, open-label, phase 1 study. Lancet. Oncol. 2017, 18, 42–51.
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229.
- Stinchcombe, T.E. Current Treatments for Surgically Resectable, Limited-Stage, and Extensive-Stage Small Cell Lung Cancer. Oncologist 2017, 22, 1510–1517.
- Povsic, M.; Enstone, A.; Wyn, R.; Kornalska, K.; Penrod, J.R.; Yuan, Y. Real-world effectiveness and tolerability of small-cell lung cancer (SCLC) treatments: A systematic literature review (SLR). PLoS ONE 2019, 14, e0219622.
- Carelli, S.; Zadra, G.; Vaira, V.; Falleni, M.; Bottiglieri, L.; Nosotti, M.; Di Giulio, A.M.; Gorio, A.; Bosari, S. Up-regulation of focal adhesion kinase in non-small cell lung cancer. Lung Cancer 2006, 53, 263–271.
- Dy, G.K.; Ylagan, L.; Pokharel, S.; Miller, A.; Brese, E.; Bshara, W.; Morrison, C.; Cance, W.G.; Golubovskaya, V.M. The Prognostic Significance of Focal Adhesion Kinase Expression in Stage I Non-Small-Cell Lung Cancer. J. Thorac. Oncol. 2014, 9, 1278–1284.
- Wang, C.; Yang, R.; Yue, D.; Zhang, Z. Expression of FAK and PTEN in Bronchioloalveolar Carcinoma and Lung Adenocarcinoma. Lung 2009, 187, 104–109.
- Imaizumi, M.; Nishimura, M.; Takeuchi, S.; Murase, M.; Hamaguchi, M. Role of tyrosine specific phosphorylation of cellular proteins, especially EGF receptor and p125FAK in human lung cancer cells. Lung Cancer 1997, 17, 69–84.
- Ocak, S.; Chen, H.; Callison, C.; Gonzalez, A.L.; Massion, P.P. Expression of focal adhesion kinase in small-cell lung carcinoma. Cancer 2012, 118, 1293–1301.
- Hsu, N.Y.; Chen, C.Y.; Hsu, C.P.; Lin, T.Y.; Chou, M.C.; Chiou, S.H.; Chow, K.C. Prognostic significance of expression of nm23-H1 and focal adhesion kinase in non-small cell lung cancer. Oncol. Rep. 2007, 18, 81–85.
- Ji, H.F.; Pang, D.; Fu, S.B.; Jin, Y.; Yao, L.; Qi, J.P.; Bai, J. Overexpression of focal adhesion kinase correlates with increased lymph node metastasis and poor prognosis in non-small-cell lung cancer. J. Cancer Res. Clin. Oncol. 2013, 139, 429–435.
- Owens, L.V.; Xu, L.; Craven, R.J.; Dent, G.A.; Weiner, T.M.; Kornberg, L.; Liu, E.T.; Cance, W.G. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res. 1995, 55, 2752–2755.
- Ocak, S.; Yamashita, H.; Udyavar, A.R.; Miller, A.N.; Gonzalez, A.L.; Zou, Y.; Jiang, A.; Yi, Y.; Shyr, Y.; Estrada, L.; et al. DNA copy number aberrations in small-cell lung cancer reveal activation of the focal adhesion pathway. Oncogene 2010, 29, 6331–6342.
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355.
- Schaller, M.D.; Borgman, C.A.; Parsons, J.T. Autonomous expression of a noncatalytic domain of the focal adhesion-associated protein tyrosine kinase pp125FAK. Mol. Cell. Biol. 1993, 13, 785–791.
- Richardson, A.; Parsons, T. A mechanism for regulation of the adhesion-associated proteintyrosine kinase pp125FAK. Nature 1996, 380, 538–540.
- Kleinschmidt, E.G.; Schlaepfer, D.D. Focal adhesion kinase signaling in unexpected places. Curr. Opin. Cell Biol. 2017, 45, 24–30.
- Almeida, E.A.; Ilic, D.; Han, Q.; Hauck, C.R.; Jin, F.; Kawakatsu, H.; Schlaepfer, D.D.; Damsky, C.H. Matrix survival signaling: From fibronectin via focal adhesion kinase to c-Jun NH(2)-terminal kinase. J. Cell Biol. 2000, 149, 741–754.
- Mak, G.; Soria, J.C.; Blagden, S.P.; Plummer, R.; Fleming, R.A.; Nebot, N.; Zhang, J.; Mazumdar, J.; Rogan, D.; Gazzah, A.; et al. A phase Ib dose-finding, pharmacokinetic study of the focal adhesion kinase inhibitor GSK2256098 and trametinib in patients with advanced solid tumours. Br. J. Cancer 2019, 120, 975–981.
- Soria, J.C.; Gan, H.K.; Blagden, S.P.; Plummer, R.; Arkenau, H.T.; Ranson, M.; Evans, T.R.; Zalcman, G.; Bahleda, R.; Hollebecque, A.; et al. A phase I, pharmacokinetic and pharmacodynamic study of GSK2256098, a focal adhesion kinase inhibitor, in patients with advanced solid tumors. Ann. Oncol. 2016, 27, 2268–2274.
- Brown, N.F.; Williams, M.; Arkenau, H.T.; Fleming, R.A.; Tolson, J.; Yan, L.; Zhang, J.; Swartz, L.; Singh, R.; Auger, K.R.; et al. A study of the focal adhesion kinase inhibitor GSK2256098 in patients with recurrent glioblastoma with evaluation of tumor penetration of [11C] GSK2256098. Neuro Oncol. 2018, 20, 1634–1642.
- Infante, J.R.; Camidge, D.R.; Mileshkin, L.R.; Chen, E.X.; Hicks, R.J.; Rischin, D.; Fingert, H.; Pierce, K.J.; Xu, H.; Roberts, W.G.; et al. Safety, pharmacokinetic, and pharmacodynamic phase I dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J. Clin. Oncol. 2012, 30, 1527–1533.
- Jones, S.F.; Siu, L.L.; Bendell, J.C.; Cleary, J.M.; Razak, A.R.; Infante, J.R.; Pandya, S.S.; Bedard, P.L.; Pierce, K.J.; Houk, B.; et al. A phase I study of VS-6063, a second-generation focal adhesion kinase inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 1100–1107.
- Patel, M.R.; Infante, J.R.; Moore, K.N.; Keegan, M.; Poli, A.; Padval, M.; Jones, S.F.; Horobin, J.; Burris, H.A. Phase 1/1b study of the FAK inhibitor defactinib (VS-6063) in combination with weekly paclitaxel for advanced ovarian cancer. J. Clin. Oncol. 2014, 32, 5521.
- Shimizu, T.; Fukuoka, K.; Takeda, M.; Iwasa, T.; Yoshida, T.; Horobin, J.; Keegan, M.; Vaickus, L.; Chavan, A.; Padval, M.; et al. A first-in-Asian phase 1 study to evaluate safety, pharmacokinetics and clinical activity of VS-6063, a focal adhesion kinase (FAK) inhibitor in Japanese patients with advanced solid tumors. Cancer Chemother. Pharm. 2016, 77, 997–1003.
- Doi, T.; Yang, J.C.; Shitara, K.; Naito, Y.; Cheng, A.L.; Sarashina, A.; Pronk, L.C.; Takeuchi, Y.; Lin, C.C. Phase I Study of the Focal Adhesion Kinase Inhibitor BI 853520 in Japanese and Taiwanese Patients with Advanced or Metastatic Solid Tumors. Target Oncol. 2019, 14, 57–65.
- De Jonge, M.J.A.; Steeghs, N.; Lolkema, M.P.; Hotte, S.J.; Hirte, H.W.; van der Biessen, D.A.J.; Abdul Razak, A.R.; De Vos, F.; Verheijen, R.B.; Schnell, D.; et al. Phase I Study of BI 853520, an Inhibitor of Focal Adhesion Kinase, in Patients with Advanced or Metastatic Nonhematologic Malignancies. Target Oncol. 2019, 14, 43–55.
- Verheijen, R.B.; van der Biessen, D.A.J.; Hotte, S.J.; Siu, L.L.; Spreafico, A.; de Jonge, M.J.A.; Pronk, L.C.; De Vos, F.; Schnell, D.; Hirte, H.W.; et al. Randomized, Open-Label, Crossover Studies Evaluating the Effect of Food and Liquid Formulation on the Pharmacokinetics of the Novel Focal Adhesion Kinase (FAK) Inhibitor BI 853520. Target Oncol. 2019, 14, 67–74.
- Aung, K.L.; McWhirter, E.; Welch, S.; Wang, L.; Lovell, S.; Stayner, L.-A.; Ali, S.; Malpage, A.; Makepeace, B.; Ramachandran, M.; et al. A phase II trial of GSK2256098 and trametinib in patients with advanced pancreatic ductal adenocarcinoma (PDAC) (MOBILITY-002 Trial, NCT02428270). J. Clin. Oncol. 2018, 36, 409.
- Fennell, D.A.; Baas, P.; Taylor, P.; Nowak, A.K.; Gilligan, D.; Nakano, T.; Pachter, J.A.; Weaver, D.T.; Scherpereel, A.; Pavlakis, N.; et al. Maintenance Defactinib Versus Placebo After First-Line Chemotherapy in Patients With Merlin-Stratified Pleural Mesothelioma: COMMAND—A Double-Blind, Randomized, Phase II Study. J. Clin. Oncol. 2019, 37, 790–798.
- Bueno, R.; Gill, R.R.; Lizotte, P.H.; Sprott, K.; Jackman, D.M.; Barlow, J.; Sharma, S.; Yeap, B.Y.; Chirieac, L.R.; Lebenthal, A.; et al. Effect of FAK inhibitor defactinib on tumor immune changes and tumor reductions in a phase II window of opportunity study in malignant pleural mesothelioma (MPM). J. Clin. Oncol. 2017, 35, 8555.
- Liu, T.-J.; LaFortune, T.; Honda, T.; Ohmori, O.; Hatakeyama, S.; Meyer, T.; Jackson, D.; de Groot, J.; Yung, W.K.A. Inhibition of both focal adhesion kinase and insulin-like growth factor-I receptor kinase suppresses glioma proliferation in vitro and in vivo. Mol. Cancer Ther. 2007, 6, 1357–1367.
- Slack-Davis, J.K.; Martin, K.H.; Tilghman, R.W.; Iwanicki, M.; Ung, E.J.; Autry, C.; Luzzio, M.J.; Cooper, B.; Kath, J.C.; Roberts, W.G.; et al. Cellular Characterization of a Novel Focal Adhesion Kinase Inhibitor. J. Biol. Chem. 2007, 282, 14845–14852.
- Auger, K.R.; Smitheman, K.N.; Korenchuk, S.; McHugh, C.; Kruger, R.; Van Aller, G.S.; Smallwood, A.; Gontarek, R.R.; Faitg, T.; Johnson, N. 387 The Focal Adhesion Kinase Inhibitor GSK2256098: A Potent and Selective Inhibitor for the Treatment of Cancer. Eur. J. Cancer 2012, 48, 118.
- Weis, S.M.; Lim, S.T.; Lutu-Fuga, K.M.; Barnes, L.A.; Chen, X.L.; Gothert, J.R.; Shen, T.L.; Guan, J.L.; Schlaepfer, D.D.; Cheresh, D.A. Compensatory role for Pyk2 during angiogenesis in adult mice lacking endothelial cell FAK. J. Cell Biol. 2008, 181, 43–50.
- Walsh, C.; Tanjoni, I.; Uryu, S.; Tomar, A.; Nam, J.O.; Luo, H.; Phillips, A.; Patel, N.; Kwok, C.; McMahon, G.; et al. Oral delivery of PND-1186 FAK inhibitor decreases tumor growth and spontaneous breast to lung metastasis in pre-clinical models. Cancer Biol. 2010, 9, 778–790.
- Roberts, W.G.; Ung, E.; Whalen, P.; Cooper, B.; Hulford, C.; Autry, C.; Richter, D.; Emerson, E.; Lin, J.; Kath, J.; et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008, 68, 1935–1944.
- Kang, Y.; Hu, W.; Ivan, C.; Dalton, H.J.; Miyake, T.; Pecot, C.V.; Zand, B.; Liu, T.; Huang, J.; Jennings, N.B.; et al. Role of focal adhesion kinase in regulating YB-1-mediated paclitaxel resistance in ovarian cancer. J. Natl. Cancer Inst. 2013, 105, 1485–1495.
- Heinrich, T.; Seenisamy, J.; Emmanuvel, L.; Kulkarni, S.S.; Bomke, J.; Rohdich, F.; Greiner, H.; Esdar, C.; Krier, M.; Grädler, U.; et al. Fragment-Based Discovery of New Highly Substituted 1H-Pyrrolo[2,3-b]- and 3H-Imidazolo[4,5-b]-Pyridines as Focal Adhesion Kinase Inhibitors. J. Med. Chem. 2013, 56, 1160–1170.
- Kurenova, E.; Liao, J.; He, D.H.; Hunt, D.; Yemma, M.; Bshara, W.; Seshadri, M.; Cance, W.G. The FAK scaffold inhibitor C4 disrupts FAK-VEGFR-3 signaling and inhibits pancreatic cancer growth. Oncotarget 2013, 4, 1632–1646.
- Kurenova, E.V.; Hunt, D.L.; He, D.; Magis, A.T.; Ostrov, D.A.; Cance, W.G. Small molecule chloropyramine hydrochloride (C4) targets the binding site of focal adhesion kinase and vascular endothelial growth factor receptor 3 and suppresses breast cancer growth in vivo. J. Med. Chem. 2009, 52, 4716–4724.
- Stewart, J.E.; Ma, X.; Megison, M.; Nabers, H.; Cance, W.G.; Kurenova, E.V.; Beierle, E.A. Inhibition of FAK and VEGFR-3 binding decreases tumorigenicity in neuroblastoma. Mol. Carcinog. 2015, 54, 9–23.
- Golubovskaya, V.M.; Ho, B.; Zheng, M.; Magis, A.; Ostrov, D.; Morrison, C.; Cance, W.G. Disruption of focal adhesion kinase and p53 interaction with small molecule compound R2 reactivated p53 and blocked tumor growth. BMC Cancer 2013, 13, 342.
- Ho, B.; Olson, G.; Figel, S.; Gelman, I.; Cance, W.G.; Golubovskaya, V.M. Nanog increases focal adhesion kinase (FAK) promoter activity and expression and directly binds to FAK protein to be phosphorylated. J. Biol. Chem. 2012, 287, 18656–18673.
- Tiede, S.; Meyer-Schaller, N.; Kalathur, R.K.R.; Ivanek, R.; Fagiani, E.; Schmassmann, P.; Stillhard, P.; Hafliger, S.; Kraut, N.; Schweifer, N.; et al. The FAK inhibitor BI 853520 exerts anti-tumor effects in breast cancer. Oncogenesis 2018, 7, 73.
- Laszlo, V.; Valko, Z.; Ozsvar, J.; Kovacs, I.; Garay, T.; Hoda, M.A.; Klikovits, T.; Stockhammer, P.; Aigner, C.; Groger, M.; et al. The FAK inhibitor BI 853520 inhibits spheroid formation and orthotopic tumor growth in malignant pleural mesothelioma. J. Mol. Med. 2019, 97, 231–242.
- Shi, Q.; Hjelmeland, A.B.; Keir, S.T.; Song, L.; Wickman, S.; Jackson, D.; Ohmori, O.; Bigner, D.D.; Friedman, H.S.; Rich, J.N. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Mol. Carcinog. 2007, 46, 488–496.
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretic, L.; Kong, G.; Leenders, F.; Lu, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53.
- Tallett, A.; Chilvers, E.R.; MacKinnon, A.C.; Haslett, C.; Sethi, T. Neuropeptides stimulate tyrosine phosphorylation and tyrosine kinase activity in small cell lung cancer cell lines. Peptides 1996, 17, 665–673.
- Frisch, S.M.; Vuori, K.; Ruoslahti, E.; Chan-Hui, P.Y. Control of adhesion-dependent cell survival by focal adhesion kinase. J. Cell Biol. 1996, 134, 793–799.
- Kurenova, E.; Xu, L.H.; Yang, X.; Baldwin, A.S.; Craven, R.J.; Hanks, S.K.; Liu, Z.G.; Cance, W.G. Focal adhesion kinase suppresses apoptosis by binding to the death domain of receptor-interacting protein. Mol. Cell. Biol. 2004, 24, 4361–4371.
- Schlaepfer, D.D.; Hanks, S.K.; Hunter, T.; van der Geer, P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature 1994, 372, 786–791.
- Oktay, M.; Wary, K.K.; Dans, M.; Birge, R.B.; Giancotti, F.G. Integrin-mediated activation of focal adhesion kinase is required for signaling to Jun NH2-terminal kinase and progression through the G1 phase of the cell cycle. J. Cell Biol. 1999, 145, 1461–1469.
- Zhao, J.; Bian, Z.C.; Yee, K.; Chen, B.P.; Chien, S.; Guan, J.L. Identification of transcription factor KLF8 as a downstream target of focal adhesion kinase in its regulation of cyclin D1 and cell cycle progression. Mol. Cell 2003, 11, 1503–1515.
- Zhao, J.; Pestell, R.; Guan, J.L. Transcriptional activation of cyclin D1 promoter by FAK contributes to cell cycle progression. Mol. Biol. Cell 2001, 12, 4066–4077.
- Zhao, J.H.; Reiske, H.; Guan, J.L. Regulation of the cell cycle by focal adhesion kinase. J. Cell Biol. 1998, 143, 1997–2008.
- Bond, M.; Sala-Newby, G.B.; Newby, A.C. Focal adhesion kinase (FAK)-dependent regulation of S-phase kinase-associated protein-2 (Skp-2) stability. A novel mechanism regulating smooth muscle cell proliferation. J. Biol. Chem. 2004, 279, 37304–37310.
- Bryant, P.; Zheng, Q.; Pumiglia, K. Focal adhesion kinase controls cellular levels of p27/Kip1 and p21/Cip1 through Skp2-dependent and -independent mechanisms. Mol. Cell. Biol. 2006, 26, 4201–4213.
- Carrano, A.C.; Eytan, E.; Hershko, A.; Pagano, M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol. 1999, 1, 193–199.
- Aboubakar Nana, F.; Lecocq, M.; Ladjemi, M.Z.; Detry, B.; Dupasquier, S.; Feron, O.; Massion, P.P.; Sibille, Y.; Pilette, C.; Ocak, S. Therapeutic Potential of Focal Adhesion Kinase Inhibition in Small Cell Lung Cancer. Mol. Cancer 2019, 18, 17–27.
- Ding, Q.; Grammer, J.R.; Nelson, M.A.; Guan, J.L.; Stewart, J.E.; Gladson, C.L. p27Kip1 and cyclin D1 are necessary for focal adhesion kinase regulation of cell cycle progression in glioblastoma cells propagated in vitro and in vivo in the scid mouse brain. J. Biol. Chem. 2005, 280, 6802–6815.
- Walker, J.L.; Fournier, A.K.; Assoian, R.K. Regulation of growth factor signaling and cell cycle progression by cell adhesion and adhesion-dependent changes in cellular tension. Cytokine Growth Factor Rev. 2005, 16, 395–405.
- Haskell, H.; Natarajan, M.; Hecker, T.P.; Ding, Q.; Stewart, J.; Grammer, J.R.; Gladson, C.L. Focal adhesion kinase is expressed in the angiogenic blood vessels of malignant astrocytic tumors in vivo and promotes capillary tube formation of brain microvascular endothelial cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 2157–2165.
- Hauck, C.R.; Hsia, D.A.; Ilic, D.; Schlaepfer, D.D. v-Src SH3-enhanced interaction with focal adhesion kinase at beta 1 integrin-containing invadopodia promotes cell invasion. J. Biol. Chem. 2002, 277, 12487–12490.
- Hsia, D.A.; Mitra, S.K.; Hauck, C.R.; Streblow, D.N.; Nelson, J.A.; Ilic, D.; Huang, S.; Li, E.; Nemerow, G.R.; Leng, J.; et al. Differential regulation of cell motility and invasion by FAK. J. Cell Biol. 2003, 160, 753–767.
- Kornberg, L.J. Focal adhesion kinase and its potential involvement in tumor invasion and metastasis. Head Neck 1998, 20, 745–752.
- Burridge, K.; Turner, C.E.; Romer, L.H. Tyrosine phosphorylation of paxillin and pp125FAK accompanies cell adhesion to extracellular matrix: A role in cytoskeletal assembly. J. Cell Biol. 1992, 119, 893–903.
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709.
- Sheetz, M.P.; Felsenfeld, D.P.; Galbraith, C.G. Cell migration: Regulation of force on extracellular-matrix-integrin complexes. Trends Cell. Biol. 1998, 8, 51–54.
- Pelham, R.J.; Wang, Y. High resolution detection of mechanical forces exerted by locomoting fibroblasts on the substrate. Mol. Biol. Cell. 1999, 10, 935–945.
- Katsumi, A.; Orr, A.W.; Tzima, E.; Schwartz, M.A. Integrins in mechanotransduction. J. Biol. Chem. 2004, 279, 12001–12004.
- Hanks, S.K.; Ryzhova, L.; Shin, N.Y.; Brabek, J. Focal adhesion kinase signaling activities and their implications in the control of cell survival and motility. Front. Biosci. A J. Virtual Libr. 2003, 8, 982–996.
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell. Biol. 2005, 6, 56–68.
- Parsons, J.T. Focal adhesion kinase: The first ten years. J. Cell Sci. 2003, 116, 1409–1416.
- Siesser, P.M.; Hanks, S.K. The signaling and biological implications of FAK overexpression in cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 3233–3237.
- Webb, D.J.; Donais, K.; Whitmore, L.A.; Thomas, S.M.; Turner, C.E.; Parsons, J.T.; Horwitz, A.F. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 2004, 6, 154–161.
- Tilghman, R.W.; Slack-Davis, J.K.; Sergina, N.; Martin, K.H.; Iwanicki, M.; Hershey, E.D.; Beggs, H.E.; Reichardt, L.F.; Parsons, J.T. Focal adhesion kinase is required for the spatial organization of the leading edge in migrating cells. J. Cell Sci. 2005, 118, 2613–2623.
- Klemke, R.L.; Leng, J.; Molander, R.; Brooks, P.C.; Vuori, K.; Cheresh, D.A. CAS/Crk coupling serves as a “molecular switch” for induction of cell migration. J. Cell Biol. 1998, 140, 961–972.
- Cary, L.A.; Chang, J.F.; Guan, J.L. Stimulation of cell migration by overexpression of focal adhesion kinase and its association with Src and Fyn. J. Cell Sci. 1996, 109, 1787–1794.
- Cary, L.A.; Han, D.C.; Polte, T.R.; Hanks, S.K.; Guan, J.L. Identification of p130Cas as a mediator of focal adhesion kinase-promoted cell migration. J. Cell Biol. 1998, 140, 211–221.
- Owen, J.D.; Ruest, P.J.; Fry, D.W.; Hanks, S.K. Induced focal adhesion kinase (FAK) expression in FAK-null cells enhances cell spreading and migration requiring both auto- and activation loop phosphorylation sites and inhibits adhesion-dependent tyrosine phosphorylation of Pyk2. Mol. Cell. Biol. 1999, 19, 4806–4818.
- Sieg, D.J.; Hauck, C.R.; Schlaepfer, D.D. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J. Cell Sci. 1999, 112, 2677–2691.
- Han, D.C.; Guan, J.L. Association of focal adhesion kinase with Grb7 and its role in cell migration. J. Biol. Chem. 1999, 274, 24425–24430.
- Han, D.C.; Shen, T.L.; Guan, J.L. Role of Grb7 targeting to focal contacts and its phosphorylation by focal adhesion kinase in regulation of cell migration. J. Biol. Chem. 2000, 275, 28911–28917.
- Irby, R.B.; Yeatman, T.J. Increased Src activity disrupts cadherin/catenin-mediated homotypic adhesion in human colon cancer and transformed rodent cells. Cancer Res. 2002, 62, 2669–2674.
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell. Biol. 2006, 7, 131–142.
- Avizienyte, E.; Frame, M.C. Src and FAK signalling controls adhesion fate and the epithelial-to-mesenchymal transition. Curr. Opin. Cell Biol. 2005, 17, 542–547.
- McLean, G.W.; Carragher, N.O.; Avizienyte, E.; Evans, J.; Brunton, V.G.; Frame, M.C. The role of focal-adhesion kinase in cancer—A new therapeutic opportunity. Nat. Rev. Cancer 2005, 5, 505–515.
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374.
- Canadas, I.; Taus, A.; Gonzalez, I.; Villanueva, X.; Gimeno, J.; Pijuan, L.; Domine, M.; Sanchez-Font, A.; Vollmer, I.; Menendez, S.; et al. High circulating hepatocyte growth factor levels associate with epithelial to mesenchymal transition and poor outcome in small cell lung cancer patients. Oncotarget 2014, 5, 5246–5256.
- Ito, T.; Kudoh, S.; Ichimura, T.; Fujino, K.; Hassan, W.A.; Udaka, N. Small cell lung cancer, an epithelial to mesenchymal transition (EMT)-like cancer: Significance of inactive Notch signaling and expression of achaete-scute complex homologue 1. Hum. Cell 2017, 30, 1–10.
- Zhao, L.; Li, J.; Liu, Y.; Zhou, W.; Shan, Y.; Fan, X.; Zhou, X.; Shan, B.; Song, Y.; Zhan, Q. Flotillin1 promotes EMT of human small cell lung cancer via TGF-beta signaling pathway. Cancer Biol. Med. 2018, 15, 400–414.
- Cicchini, C.; Laudadio, I.; Citarella, F.; Corazzari, M.; Steindler, C.; Conigliaro, A.; Fantoni, A.; Amicone, L.; Tripodi, M. TGFbeta-induced EMT requires focal adhesion kinase (FAK) signaling. Exp. Cell Res. 2008, 314, 143–152.
- Fan, H.; Zhao, X.; Sun, S.; Luo, M.; Guan, J.L. Function of focal adhesion kinase scaffolding to mediate endophilin A2 phosphorylation promotes epithelial-mesenchymal transition and mammary cancer stem cell activities in vivo. J. Biol. Chem. 2013, 288, 3322–3333.
- Li, X.Y.; Zhou, X.; Rowe, R.G.; Hu, Y.; Schlaepfer, D.D.; Ilic, D.; Dressler, G.; Park, A.; Guan, J.L.; Weiss, S.J. Snail1 controls epithelial-mesenchymal lineage commitment in focal adhesion kinase-null embryonic cells. J. Cell Biol. 2011, 195, 729–738.
- Wang, X.; Urvalek, A.M.; Liu, J.; Zhao, J. Activation of KLF8 transcription by focal adhesion kinase in human ovarian epithelial and cancer cells. J. Biol. Chem. 2008, 283, 13934–13942.
- Wang, X.; Zheng, M.; Liu, G.; Xia, W.; McKeown-Longo, P.J.; Hung, M.C.; Zhao, J. Kruppel-like factor 8 induces epithelial to mesenchymal transition and epithelial cell invasion. Cancer Res. 2007, 67, 7184–7193.
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; De Herreros, A.G. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84.
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor Snail controls epithelial—Mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83.
- Liu, S.Q.; Xu, C.Y.; Wu, W.H.; Fu, Z.H.; He, S.W.; Qin, M.B.; Huang, J.A. Sphingosine kinase 1 promotes the metastasis of colorectal cancer by inducing the epithelialmesenchymal transition mediated by the FAK/AKT/MMPs axis. Int. J. Oncol. 2019, 54, 41–52.
More
Information
Subjects:
Medicine, General & Internal
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
19 Aug 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No