 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sang Ryong Kim | + 2907 word(s) | 2907 | 2021-08-03 07:57:10 | | | |
| 2 | Ron Wang | + 407 word(s) | 3314 | 2021-08-17 11:56:49 | | |
Video Upload Options
Reactive oxygen species (ROS) result from normal daily cellular metabolism. Research conducted in the last two decades has clarified the role of ROS as secondary signaling molecules that regulate various biological and physiological processes, including proliferation, host defense, and gene expression. Furthermore, earlier reports have also indicated the role of ROS as a signal transduction mechanism.
1. Introduction
Reactive oxygen species (ROS) result from normal daily cellular metabolism. Research conducted in the last two decades has clarified the role of ROS as secondary signaling molecules that regulate various biological and physiological processes, including proliferation, host defense, and gene expression [1][2]. Furthermore, earlier reports have also indicated the role of ROS as a signal transduction mechanism. This allows adaptation to changes in environmental nutrients and the oxidative environment [3]. In this respect, Kiley and Storz [4] have well-defined, in the prokaryotes, mechanisms whereby ROS directly activate transcription factors (TFs) for stress adaptation. On the contrary, oxidative stress (OS) refers to elevated levels of intracellular ROS, such as superoxide anion (O2•−), hydroxyl radical (OH•), and non-radical molecules, such as hydrogen peroxide (H2O2) and singlet oxygen (1O2), which further damage lipids, proteins, and DNA (Figure 1 A). A high-energy exposure or electron transfer reaction leads to the production of highly reactive ROS, which is a stepwise reduction of molecular oxygen (O2) as represented in equation (1). Moreover, ROS generation occurs at elevated rates in normal aging. It is an inevitable process in both acute and chronic pathophysiological conditions [5]. Thus, OS is usually the result of excessive ROS production, mitochondrial dysfunction, and an impaired antioxidant system, or a combination of these factors. O2 → O2•− → H2O2 → OH • → H2O (1)
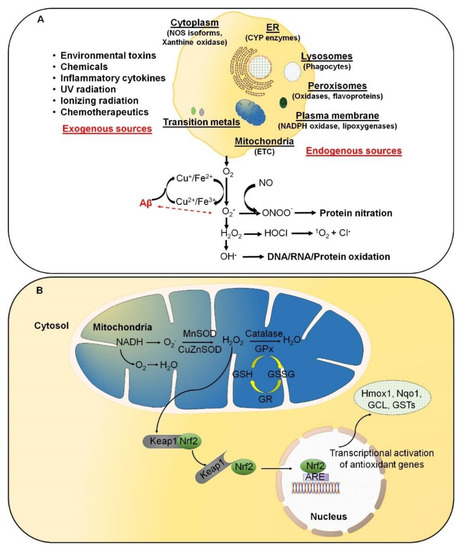
ROS are predominantly produced in mitochondria via mitochondrial enzymes. The electron transport chain (ETC) of mitochondria produces superoxide radicals at respiratory complexes I and III of the oxidative phosphorylation (OXPHOS) pathway through the single-electron leak [2][6]. Nevertheless, the ROS production rate in complex I is much less than the Flavin-dependent enzymes in the mitochondrial matrix [7]. Amongst various intracellular antioxidant enzymes, five have been mainly discussed in physiological conditions, i.e., (i) Cu/Zn-superoxide dismutase (Cu/Zn-SOD, SOD1) in the cytosol, (ii) manganese superoxide dismutase (Mn-SOD, SOD2) in the mitochondrial matrix, (iii) catalase (CAT), (iv) glutathione peroxidase (GPx), and (v) glutathione reductase. In Figure 1 B, SOD converts superoxide to O2 and H2O2, whereas CAT and GPx convert H2O2 into H2O and O2. Along with the primary antioxidant defense against ROS, secondary antioxidant and cellular detoxification programs are mainly regulated by NF-E2-related factor 2 (Nrf2) and Kelch-like ECH-associated protein 1 (Keap1). Under normal conditions, Nrf2 is retained in the cytoplasm by the actin-binding protein Keap1; a substrate adaptor protein for the Cullin3-containing E3–ligase complex, which targets Nrf2 for ubiquitination and degradation by the proteasome [8]. Keap1 is redox-sensitive since this protein can be modified by different oxidants and electrophiles [9]. OS abrogates the Keap1-mediated degradation of Nrf2, which in turn accumulates in the nucleus [10]. It heterodimerizes with a small musculoaponeurotic fibrosarcoma (Maf) protein on antioxidant response elements (AREs). Nrf2, along with ARE, further stimulates the expression of a wide array of phase II antioxidant enzymes, which includes NAD(P)H quinone oxidoreductase 1 (Nqo1), heme oxygenase 1 (Hmox1), glutamate-cysteine ligase, and glutathione S transferases (GSTs) [10][11][12]. In addition, Nrf2 also contributes to cellular proteostasis by regulating the expression of molecular chaperones and various proteasomal subunits [13][14][15]. Apart from antioxidant enzymes, small molecular weight and nonenzymatic antioxidants, such as vitamins, carotenoids, thiol antioxidants, and natural flavonoids, also protect intracellular components against ROS [16].
Aggregated proteins' deposition and spreading are the main characteristics of sporadic (s) and familial (f) forms of various neurodegenerative disorders, such as AD. This, in turn, results in excessive ROS production leading to OS, chronic neuroinflammation, and mitochondrial dysfunction, which altogether cause neuronal loss [17] and protein misfolding [18]. ROS-induced protein misfolding/unfolding can result in gain/loss-of-function. The protein modification of the oxidized proteins is insufficient to achieve their actual shape, impacting stability, activity, and/or function [19][20]. Several lines of evidence suggest that elevated ROS production initiates toxic amyloid-beta precursor protein (APP) processing and thereby triggers amyloid-beta (Aβ) generation [21][22]. These elevations in ROS are the results of protein aggregation and corresponding neuronal damage, which in turn activates disease-associated microglia via damage-associated molecular patterns [23]. These ROS are primarily generated via NADPH oxidase 2, which is well associated with DAMP signaling, inflammation, and amyloid plaque deposition [23]. Additionally, ROS generated from mitochondria helps in the propagation of immune activation, leading to excessive OS and neurodegeneration. Interestingly, recent studies on postmortem AD brains and AD transgenic mice have shown that Aβ and APP are found in mitochondrial membranes to block protein transport and disrupt the ETC with final, irreversible cell damage [24]. Moreover, these disruptions are further exacerbated by a defective repair system. Tamagno and colleagues reported that OS resulting from hydroxynonenal (HNE) or H2O2 leads to enhanced Aβ production in different cell models [21]. In addition, HNE also modifies the γ-secretase substrate receptor nicastrin, which leads to enhanced binding of the γ-secretase substrate APP and likely results in elevated Aβ generation [22]. Moreover, neurons contain a high amount of polyunsaturated fatty acids (PUFAs) that can interact with ROS, leading to a self-propagating cascade of lipid peroxidation and molecular destruction [25]. Products of lipid peroxidation have also been shown to be elevated in blood samples and brains of AD patients at autopsy [26][27]. Both nuclear and mitochondrial DNA and RNA also exhibit oxidative damage in the AD brain [28][29][30]. Hence, understanding oxidative balance is regarded as an important event in understanding AD pathogenesis. OS might increase the aggregation and production of Aβ and assist polymerization and tau phosphorylation via the creation of a vicious cycle that stimulates the progression and even initiation of AD. Keeping this in mind, in this review, we sought to analyze the myriad interactions between oxygen radicals and toxic protein oligomers in the context of AD to understand their importance in disease pathogenesis. Furthermore, we also discuss the role of microbiota in altering the redox balance and its consequences concerning Aβ production and tau hyperphosphorylation.
2. Markers of Oxidative Stress
ROS are oxygen-containing molecules that are chemically more reactive than O2 and, therefore, can damage cellular macromolecules. For example, ROS can react with nucleic acids (NA) by attacking nitrogenous bases and the sugar-phosphate backbone. Further, these can evoke single- and double-stranded DNA breaks, affecting the protein-coding region of mtDNA and influencing OXPHOS [31][32]. mtDNA mutations can cause disturbances in the respiratory chain, and as a result, it loses control over ROS production [1]. In addition, the modification in core DNA repair genes can result in an impaired recognition system and an inefficient repair of DNA damage, which in turn can accelerate aging and leads to age-related disruptions in cellular and tissue functions. This also results in the accumulation of ROS, which increases with age and intensifies OS. This elevation in OS damages mtDNA, leading to apoptosis, inhibition of mitochondrial respiratory chain transition, and increased mitochondrial membrane permeability in the absence of sufficient antioxidant capacity [5]. Thus, pro-oxidative/antioxidative cellular imbalance between ROS production and the ability of the defense mechanisms of biological systems to eliminate ROS-mediated cellular stress disturbances results in a vicious cycle, since the OS reciprocally aggravates ROS production. ROS have also been reported to attack structural and enzymatic proteins via oxidation of residual amino acids, prosthetic groups, formation of cross-links and protein aggregates, and proteolysis [32]. Lipid peroxidation (auto-oxidation) is a process in which PUFAs are oxidized due to several double bonds in their structure. This process involves producing peroxides (chemical compounds in which a single covalent bond links two oxygen atoms), ROS, and other reactive organic free radicals. Several markers of oxidative damage have been defined, including the following: 8-hydroxy-2-deoxyguanosine (8-OHdG) and 8-hydroxyguanosine, markers of oxidative DNA damage; 8-hydroxyguanine, a marker of RNA oxidation; protein carbonyls and nitrotyrosine, markers of protein oxidation; and malondialdehyde (MDA), thiobarbituric-acid-reactive substances, 4-hydroxy-2-nonenal (4-HNE), acrolein, isoprostanes, and neuroprostanes, markers of lipid peroxidation [5][32][33]. Moreover, ROS and aging have also been linked to the promotion and accumulation of advanced glycation end products (AGEs). AGEs are insoluble in detergents, protease-resistant, and non-degradable protein, lipid, or NA aggregates generated by non-enzymatic glycation or glycoxidation after exposure to aldose sugar. AGEs have been reported to impair normal cellular/tissue functions directly or indirectly through the AGE/RAGE pathway after binding to specific receptors for advanced glycation end products (RAGEs) [34]. Due to synergism with OS, the production of AGEs is promoted by OS, which eventually leads to ROS generation.
Furthermore, AGEs have been found to accumulate in numerous tissues throughout physiological aging, which leads to OS since the ability to respond to OS reduces with age. Due to this, many proteins lose their function, including those involved in gene transcription regulation [32][33]. Thus, AGEs serve not only as proinflammatory molecules but also as potent neurotoxins [35]. Protein glycation begins as a nonenzymatic process with a free amino acid group capable of producing a labile Schiff base. The process thus takes place along with the unconstrained condensation of aldehyde or ketone groups reportedly present in sugars. Furthermore, the phenomenon mentioned above also agrees with Maillard’s classical reaction in 1912 [36][37]. Subsequently, a series of reactions occur that result in the generation of AGEs containing irreversibly cross-linked heterogeneous protein aggregates.
3. Linking Microbiota with Oxidative Stress and AD
Recently, several pieces of evidence link the role of microbiota in brain biology and aging, being an essential factor involved in various physiological processes via interactive symbiotic network system with host [38][39][40][41][42][43]. This interactive network between host and microbiota interconnects the gut track, epidermis, liver, and all other organs with the central nervous system generally referred to as the microbiota-gut-brain axis [44][45]. The microbiota is composed mainly of bacteria that colonize all mucosal surfaces, with higher density in the gastrointestinal tract, approximately 100 trillion bacteria from nearly 1000 various bacterial species [38][44], thereby influencing and triggering various events associated with aging disorders such as AD [38][39][40][44][46]. Recently, a line of evidence revealed an association of brain amyloidosis with pro-inflammatory gut bacteria in cognitively impaired patients [47] and various AD mouse models [48][49]. These findings strongly highlight the association of microbiota and amyloid pathogenesis in AD. However, these fields lack crucial in-depth information and require more exploration.
OS's physiological levels have been generated in the microbiota, which can interfere with its composition and functionality [50]. Furthermore, interactions between microbe-microbe or host-microbiota may also impact the CNS redox balance by elevating ROS levels or impairing the antioxidant system or both [51][52]; hence, serving not only as a cause but also a consequence of increased levels of oxidative injury in CNS [51], thus adding a new dimension to the interplay between the gut microbiota and the brain. Moreover, the microbiota can also produce a considerable amount of CNS neurotransmitters, including dopamine, serotonin, and gamma-aminobutyric acid, that can modulate the local activity of the enteric nervous system and can correlate with their respective levels within the CNS, which in turn depends on the intestinal and BBB permeability [53]. The microbiota may also produce neurotoxic and potentially neurotoxic substances (such as lipopolysaccharides and amyloid proteins), which can also reach to CNS via the systemic circulation or the vagus nerve, promoting microglial activation and neuroinflammation, elevated ROS levels, and/or making neurons more susceptible to OS [53]. Therefore, gut microbes were considered plausible triggering factors for several neurodegenerative disorders, considering the proximity of enteric nervous system neurons to the intestinal lumen [54].
However, the production of amyloid proteins helps in the formation of bacterial biofilms by promoting the binding of bacterial cells with each other, thus providing resistance from physical or immune factor-mediated destruction [46]. However, in abnormal physiological conditions, bacterial amyloids may act as prion proteins and result in cross-seeding of amyloidogenic protein that elevates pathogenic Aβ formation both in vitro and in vivo [46][55][56][57][58]. For instance, the interaction of cyanobacteria with synaptic receptors such as NMDA results in upregulation of β-N-methylamino- l -alanine (BMAA), an OS-inducing neurotoxin [59][60], in AD brains. Furthermore, BMAA has been linked with protein misfolding and resulting inflammatory consequences in the AD mice model [59][60][61]. Numerous studies also suggested a link between activation of endogenous herpes simplex-1 (HSV-1) and amyloidogenesis in AD. This intimate relationship resulted in progressive neurodegeneration and cognitive impairment, contributing to AD pathogenesis [62][63][64]. A possible reason for this could be the alteration in gut dysbiosis, which results in increased gut barrier permeability, which in turn hyper activates the innate immune response that leads to systemic inflammation, thus impairing the blood-brain barrier [46], which results in neuronal injury, protein misfolding, and neurodegeneration leading to cognitive impairment [65]. In addition, overwhelmed microglial stimulation and NF-κB-mediated proinflammatory signaling and reactive oxidative and nitrosative stressors can result in neuronal and glial cell death, which can further impair phagocytosis, leading to the accumulation of Aβ42 [66][67]. C/EBPβ/AEP signaling was activated in 3xTg mice 5xFAD mice due to gut dysbiosis, resulting in Aβ aggregates, OS, and tau hyperphosphorylation [68].
Furthermore, reduction in the relative abundance of Proteobacteria and the low levels of Bifidobacteria can reduce beneficial short-chain fatty acids, leading to lipid peroxidation [69]. This, in turn, results in impaired APP processing and trafficking, thus impacting the production of Aβ. Studies conducted using germ-free mice have confirmed the impact of microbiota on microglia maturation, astrocyte activity, neuroinflammation, OS, protein misfolding, and cognitive impairment in AD pathogenesis [49]. Modifying the gut microbiota composition with food-based therapy or supplementing with probiotics may be helpful as a new preventive and therapeutic option in both in vitro and in vivo AD models and clinical trials [66][67][70][71][72][73][74].
4. Antioxidants and AD
It is now evident that Aβ and tau pathologies are modulated by ROS and are also self-perpetuating concerning ROS formation [75]. Hence, strategies involving inhibition of Aβ oligomerization or decreasing ROS production through the design of multitargeted compounds, such as antioxidants, have resulted in several promising approaches currently being tested in clinical trials. Antioxidants are a broad and heterogeneous collection of chemicals that work by inhibiting the production, detoxification, or scavenging of oxidant species. According to a different criterion, antioxidants can be classified into four different classes based on their chemical structure: vitamins (e.g., ascorbic acid, α-tocopherol, β-carotene, and retinol), synthetic compounds (e.g., butylated hydroxytoluene), natural compounds (e.g., plant-derived polyphenols), and inorganic compounds. Some antioxidants act as chain-breaking molecules, as they can prevent the propagation of or stop radical chain reactions (e.g., α-tocopherol). On the contrary, antioxidants, such as Gpx and catalase, can detoxify H2O2. This chemical reaction serves a vital role in cell biology as H2O2 can produce OH radicals in the presence of transition metals such as Fe2+, for which there is no detoxification system [32].
Several antioxidant studies in AD models have also been reported, demonstrating that antioxidants consistently positively affect the animals’ behavioral and amyloidotic phenotypes (Table 1). Vitamins are potent antioxidants that directly affect free radicals by reducing OS, inflammatory processes, and neuronal loss [76]. Vitamin A (retinol) is essential for the neuronal formation and remains present in the nervous system across life. Along with β-carotene, vitamin A also protects regenerating neurons during the neurodegeneration process by preventing the development and aggregation of Aβ plaques both ex vivo and in vivo. It may also prevent impaired cognition in AD and improve memory performance and spatial learning in rodent models. Studies have shown that AD patients have lower vitamin A and β-carotene compared with healthy individuals [76][77]. Early vitamin E (α-tocopherol) supplementation significantly reduced Aβ levels and deposition in the Tg2576 AD model [78]. The same therapeutic regimen prevents a surge in amyloidosis [78]. It improves cognitive function after experimental traumatic brain injury, a known risk factor for AD development in Tg2576 mice [79]. Curcumin, a popular antioxidant and anti-inflammatory substance found in curry spices, substantially decreases OS and amyloid pathology in the Tg2576 mouse model [80].
Furthermore, curcumin is a potent inhibitor of Aβ fibrillization [81] and oligomerization [82] and promotes destabilization of pre-existing Aβ deposits in both cell culture models and animal models of AD [80][81][82]. Curcumin and its derivatives also increase the uptake and clearance of Aβ by macrophages in AD patients [83]. Furthermore, using LLC-PK 1 and NRK-52E cells, Balogun and colleagues reported that curcumin upregulates Aβ-induced SOD and catalase and can further activate Nrf2 by selectively binding to Keap1 [84]. Luteolin has also been associated with activating the Nrf2 pathway, which increases endogenous antioxidative gene expression in neuronal cells [85]. Melatonin, a drug with antioxidant properties, partially inhibits the expected time-dependent elevation in Aβ levels, reduces the abnormal nitration of proteins, and increases the survival of Tg2576 mice [86]. Similarly, ferulic acid, rosmarinic acid, and nordihydroguaiaretic acid (NDGA) have also been reported to inhibit the fibrillization and/or oligomerization of Aβ into higher-order species in vitro [87][88][89][90].
| Antioxidant | Mechanism | Experimental Model | Reference |
|---|---|---|---|
| Vitamins | |||
| α-Tocopherol | Reduces Aβ and lipid peroxidation; delays development of tau pathology; reduction in learning deficits and motor weakness | Tg2576 mice | [156,157,167,168,169,170,173] |
| Ascorbic acid | Reduces Aβ oligomers, tau phosphorylation, and oxidative stress | hAPP-J20 mice | [174] |
| Retinol | Reduces MDA levels; upregulates SOD activity; reduces Aβ pathology | APPswe/ PS1M146V/ tauP301L (3 × Tg) mice; in vitro enzymatic assay and in silico modeling |
[175] |
| Naturally present | |||
| CoQ10 | Reduces MDA levels; upregulates SOD activity; reduces Aβ pathology | Tg19959 mice; APP/PS1 Tg mice | [176,177] |
| Synthetic | |||
| Mito Q | Prevents cognitive decline, oxidative stress, Aβ accumulation, synaptic loss, and caspase activation | 3 × Tg mice | [178] |
| Plant-based | |||
| Zeolite | Increases endogenous SOD; reduces Aβ levels and plaque burden | Randomized clinical trial | [179] |
| β-Carotene | Improves cognitive impairment and oxidative stress | Streptozotocin-induced AD mice model | [180] |
| Curcumin | Inhibits Aβ fibrillization and oligomerization; clearance of Aβ by macrophages; reduces Aβ40 and 42 and Aβ-derived diffusible ligands; increases Aβ-degrading enzymes; promotes destabilization | Tg2576 mice; APPSw mice; APPswe/PS1dE9 mice; in vitro enzymatic assay | [160,161,162,163,181] |
| Ferulic acid | Inhibits the fibrillization and/or oligomerization of Aβ | In vitro enzymatic assay | [167,168] |
| Rosmarinic acid | Inhibits the fibrillization and/or oligomerization of Aβ | Molecular docking analysis; Tg2576 mice; PC12 neuroblastoma | [168,169,170] |
| Nordihydroguaiaretic acid (NDGA) | Inhibits the fibrillization and/or oligomerization of Aβ | Tg2576 mice | [168] |
| Mimetic | |||
| ApoE mimetic peptide Ac-hE18A-NH2 | Reduces oxidative stress and ApoE secretion; inhibits Aβ plaque deposition | APP/PS1ΔE9 mice and U251 human astrocyte cells | [182] |
| Catalase mimetic | Protects against oxidative stress, DNA, and protein oxidation; reduces Aβ and tau phosphorylation | 3 × Tg-AD mice | [183] |
| Drug | |||
| Melatonin | Inhibits time-dependent elevation of Aβ; reduces abnormal oxidation and nitration of proteins; increases survival; alleviates learning and memory deficits; decreases choline acetyltransferase activity and increases acetyltransferase activity; increases mitochondrial function | Tg2576 mice; APP 695 Tg mouse model; APP/PS1 mice; APPswePS1dE9 mice | [166,184,185,186,187] |
| N-Acetyl-l-cysteine | Reduces lipid peroxidation, oxidative stress, and glutathione peroxidase activity | APP/PS-1 knock-in mice | [188] |
The significant outcomes of these studies are reductions in Aβ levels, phosphorylated tau, mitochondrial dysfunction, microglial activation, enhanced synaptic activity, and amelioration of cognitive decline. These results indicate that antioxidant treatment is beneficial in reducing and/or preventing AD progression. The findings also show that combination therapy positively impacts cognitive behavior and lowers AD pathology. The positive findings of these studies are promising. However, they warrant prospective studies (e.g., antioxidant treatment of elderly individuals without AD) and clinical trials (antioxidant treatment for patients with AD). Recent work has also highlighted the importance of a healthy and detoxified innate response by consuming diet precursors and enhancing responsiveness [91]. For instance, the application of radiation health, such as UV radiation from the Sun, can prepare an individual for further UV exposure [92]. Another example includes exposure to pro-oxidants such as H2O2, which can prepare the body for subsequent pro-oxidant exposure, which is similar to the formation of antibodies in vaccines.
References
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15.
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95.
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653.
- Kiley, P.J.; Storz, G. Exploiting Thiol Modifications. PLoS Biol. 2004, 2, e400.
- Cui, H.; Kong, Y.; Zhang, H. Oxidative Stress, Mitochondrial Dysfunction, and Aging. J. Signal Transduct. 2012, 2012, 1–13.
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472.
- Quinlan, C.L.; Goncalves, R.L.S.; Hey-Mogensen, M.; Yadava, N.; Bunik, V.I.; Brand, M.D. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 2014, 289, 8312–8325.
- Sykiotis, G.P.; Bohmann, D. Stress-activated cap’n’collar transcription factors in aging and human disease. Sci. Signal. 2010, 3, re3.
- Holland, R.; Fishbein, J.C. Chemistry of the cysteine sensors in kelch-like ECH-associated protein. Antioxid. Redox Signal. 2010, 13, 1749–1761.
- Niforou, K.; Cheimonidou, C.; Trougakos, I.P. Molecular chaperones and proteostasis regulation during redox imbalance. Redox Biol. 2014, 2, 323–332.
- Giudice, A.; Arra, C.; Turco, M.C. Review of molecular mechanisms involved in the activation of the Nrf2-ARE signaling pathway by chemopreventive agents. Methods Mol. Biol. 2010, 647, 37–74.
- Copple, I.M.; Goldring, C.E.; Kitteringham, N.R.; Park, B.K. The Keap1-Nrf2 cellular defense pathway: Mechanisms of regulation and role in protection against drug-induced toxicity. Handb. Exp. Pharmacol. 2010, 196, 233–266.
- Hensen, S.M.M.; Heldens, L.; van Enckevort, C.M.W.; Van Genesen, S.T.; Pruijn, G.J.M.; Lubsen, N.H. Activation of the antioxidant response in methionine deprived human cells results in an HSF1-independent increase in HSPA1A mRNA levels. Biochimie 2013, 95, 1245–1251.
- Tsakiri, E.N.; Iliaki, K.K.; Höhn, A.; Grimm, S.; Papassideri, I.S.; Grune, T.; Trougakos, I.P. Diet-derived advanced glycation end products or lipofuscin disrupts proteostasis and reduces life span in Drosophila melanogaster. Free Radic. Biol. Med. 2013, 65, 1155–1163.
- Tsakiri, E.N.; Sykiotis, G.P.; Papassideri, I.S.; Terpos, E.; Dimopoulos, M.A.; Gorgoulis, V.G.; Bohmann, D.; Trougakos, I.P. Proteasome dysfunction in Drosophila signals to an Nrf2-dependent regulatory circuit aiming to restore proteostasis and prevent premature aging. Aging Cell 2013, 12, 802–813.
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15.
- Butterfield, D.A.; Drake, J.; Pocernich, C.; Castegna, A. Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid β-peptide. Trends Mol. Med. 2001, 7, 548–554.
- Tabner, B.J.; El-Agnaf, O.M.A.; German, M.J.; Fullwood, N.J.; Allsop, D. Protein aggregation, metals and oxidative stress in neurodegenerative diseases. Biochem. Soc. Trans. 2005, 33, 1082–1086.
- Pérez, V.I.; Buffenstein, R.; Masamsetti, V.; Leonard, S.; Salmon, A.B.; Mele, J.; Andziak, B.; Yang, T.; Edrey, Y.; Friguet, B.; et al. Protein stability and resistance to oxidative stress are determinants of longevity in the longest-living rodent, the naked mole-rat. Proc. Natl. Acad. Sci. USA 2009, 106, 3059–3064.
- Nedić, O.; Rattan, S.I.S.; Grune, T.; Trougakos, I.P. Molecular effects of advanced glycation end products on cell signalling pathways, ageing and pathophysiology. Free Radic. Res. 2013, 47, 28–38.
- Tamagno, E.; Bardini, P.; Obbili, A.; Vitali, A.; Borghi, R.; Zaccheo, D.; Pronzato, M.A.; Danni, O.; Smith, M.A.; Perry, G.; et al. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol. Dis. 2002, 10, 279–288.
- Tamagno, E.; Parola, M.; Bardini, P.; Piccini, A.; Borghi, R.; Guglielmotto, M.; Santoro, G.; Davit, A.; Danni, O.; Smith, M.A.; et al. β-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J. Neurochem. 2005, 92, 628–636.
- McLaurin, J.; Lai, A.Y. Mechanisms of amyloid-beta peptide uptake by neurons: The role of lipid rafts and lipid raft-associated proteins. Int. J. Alzheimers Dis. 2011, 2011, 548380.
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53.
- Nunomura, A.; Castellani, R.J.; Zhu, X.; Moreira, P.I.; Perry, G.; Smith, M.A. Involvement of oxidative stress in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2006, 65, 631–641.
- Jeandel, C.; Nicolas, M.B.; Dubois, F.; Nabet-Belleville, F.; Penin, F.; Cuny, G. Lipid peroxidation and free radical scavengers in Alzheimer’s disease. Gerontology 1989, 35, 275–282.
- Subbarao, K.V.; Richardson, J.S.; Ang, L.C. Autopsy Samples of Alzheimer’s Cortex Show Increased Peroxidation In Vitro. J. Neurochem. 1990, 55, 342–345.
- Mecocci, P.; MacGarvey, U.; Beal, M.F. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. 1994, 36, 747–751.
- Gabbita, S.P.; Lovell, M.A.; Markesbery, W.R. Increased nuclear DNA oxidation in the brain in Alzheimer’ s disease. J. Neurochem. 1998, 71, 2034–2040.
- Nunomura, A.; Perry, G.; Pappolla, M.A.; Wade, R.; Hirai, K.; Chiba, S.; Smith, M.A. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J. Neurosci. 1999, 19, 1959–1964.
- Evans, M.D.; Dizdaroglu, M.; Cooke, M.S. Oxidative DNA damage and disease: Induction, repair and significance. Mutat. Res. Rev. Mutat. Res. 2004, 567, 1–61.
- Höhn, A.; Tramutola, A.; Cascella, R. Proteostasis Failure in Neurodegenerative Diseases: Focus on Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 5497046.
- Praticò, D. Oxidative stress hypothesis in Alzheimer’s disease: A reappraisal. Trends Pharmacol. Sci. 2008, 29, 609–615.
- Schmidt, A.M.; Du Yan, S.; Yan, S.F.; Stern, D.M. The Biology of the Receptor for Advanced Glycation end Products and Its Ligands. Biochim. Biophys Acta 2000, 1498, 99–111.
- Gkogkolou, P.; Böhm, M. Advanced glycation end products: Keyplayers in skin aging? Dermatoendocrinology 2012, 4, 259.
- Maillard, L.C. Action of amino acids on sugars. Formation of melanoidins in a methodical way. Compte-Rendu L’academie Sci. 1912, 154, 66–68.
- Smith, M.A.; Taneda, S.; Richey, P.L.; Miyata, S.; Yan, S.D.; Stern, D.; Sayre, L.M.; Monnier, V.M.; Perry, G. Advanced Maillard reaction end products are associated with Alzheimer disease pathology. Proc. Natl. Acad. Sci. USA 1994, 91, 5710–5714.
- O’Toole, P.W.; Jeffery, I.B. Gut microbiota and aging. Science 2015, 350, 1214–1215.
- Collins, S.M.; Bercik, P. The relationship between intestinal microbiota and the central nervous system in normal gastrointestinal function and disease. Gastroenterology 2009, 136, 2003–2014.
- Collins, S.M.; Surette, M.G.; Bercik, P. The interplay between the intestinal microbiota and the brain. Nat. Rev. Microbiol. 2012, 10, 735–742.
- Cryan, J.F.; O’Mahony, S.M. The microbiome-gut-brain axis: From bowel to behavior. Neurogastroenterol. Motil. 2011, 23, 187–192.
- Belkaid, Y.; Hand, T. Role of the Microbiota in Immunity and inflammation. Cell 2014, 157, 121.
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268.
- Bhattacharjee, S.; Lukiw, W.J. Alzheimer’s disease and the microbiome. Front. Cell. Neurosci. 2013, 7, 153.
- Douglas-Escobar, M.; Elliott, E.; Neu, J. Effect of Intestinal Microbial Ecology on the Developing Brain. JAMA Pediatr. 2013, 167, 374–379.
- Kowalski, K.; Mulak, A. Brain-Gut-Microbiota Axis in Alzheimer’s Disease. J. Neurogastroenterol. Motil. 2019, 25, 48.
- Cattaneo, A.; Cattane, N.; Galluzzi, S.; Provasi, S.; Lopizzo, N.; Festari, C.; Ferrari, C.; Guerra, U.P.; Paghera, B.; Muscio, C.; et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 2017, 49, 60–68.
- Minter, M.R.; Zhang, C.; Leone, V.; Ringus, D.L.; Zhang, X.; Oyler-Castrillo, P.; Musch, M.W.; Liao, F.; Ward, J.F.; Holtzman, D.M.; et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016, 6, 30028.
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Coy, K.D.M.; Frisoni, G.; Neher, J.J.; Fåk, F.; Jucker, M.; Lasser, T.; et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, 1–15.
- Reese, A.T.; Cho, E.H.; Klitzman, B.; Nichols, S.P.; A Wisniewski, N.; Villa, M.M.; Durand, H.K.; Jiang, S.; Midani, F.S.; Nimmagadda, S.N.; et al. Antibiotic-induced changes in the microbiota disrupt redox dynamics in the gut. eLife 2018, 7, e35987.
- Mercante, J.W.; Neish, A.S. Reactive Oxygen Production Induced by the Gut Microbiota: Pharmacotherapeutic Implications. Curr. Med. Chem. 2012, 19, 1519–1529.
- Bonaz, B.; Bazin, T.; Pellissier, S. The Vagus Nerve at the Interface of the Microbiota-Gut-Brain Axis. Front. Neurosci. 2018, 12, 49.
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503.
- Friedland, R.P.; Chapman, M.R. The role of microbial amyloid in neurodegeneration. PLoS Pathog. 2017, 13, e1006654.
- Chen, S.G.; Stribinskis, V.; Rane, M.J.; Demuth, D.R.; Gozal, E.; Roberts, A.M.; Jagadapillai, R.; Liu, R.; Choe, K.; Shivakumar, B.; et al. Exposure to the Functional Bacterial Amyloid Protein Curli Enhances Alpha-Synuclein Aggregation in Aged Fischer 344 Rats and Caenorhabditis elegans. Sci. Rep. 2016, 6, 34477.
- Kahn, M.S.; Kranjac, D.; Alonzo, C.A.; Haase, J.H.; Cedillos, R.O.; McLinden, K.A.; Boehm, G.W.; Chumley, M.J. Prolonged elevation in hippocampal Aβ and cognitive deficits following repeated endotoxin exposure in the mouse. Behav. Brain Res. 2012, 229, 176–184.
- Lundmark, K.; Westermark, G.T.; Olsen, A.; Westermark, P. Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism. Proc. Natl. Acad. Sci. USA 2005, 102, 6098–6102.
- Zhou, Y.; Smith, D.; Leong, B.J.; Brännström, K.; Almqvist, F.; Chapman, M.R. Promiscuous Cross-seeding between Bacterial Amyloids Promotes Interspecies Biofilms. J. Biol. Chem. 2012, 287, 35092.
- Mitew, S.; Kirkcaldie, M.T.; Dickson, T.; Vickers, J.C. Altered synapses and gliotransmission in Alzheimer’s disease and AD model mice. Neurobiol. Aging 2013, 34, 2341–2351.
- Brenner, S.R. Blue-green algae or cyanobacteria in the intestinal micro-flora may produce neurotoxins such as Beta-N-Methylamino-l-Alanine (BMAA) which may be related to development of amyotrophic lateral sclerosis, Alzheimer’s disease and Parkinson-Dementia-Complex in humans and Equine Motor Neuron Disease in Horses. Med. Hypotheses 2013, 80, 103.
- Schwartz, K.; Boles, B.R. Microbial amyloids—Functions and interactions within the host. Curr. Opin. Microbiol. 2013, 16, 93–99.
- Hill, J.M.; Zhao, Y.; Clement, C.; Neumann, D.M.; Lukiw, W.J. HSV-1 infection of human brain cells induces miRNA-146a and Alzheimer-type inflammatory signaling. Neuroreport 2009, 20, 1500.
- Ball, M.J.; Lukiw, W.J.; Kammerman, E.M.; Hill, J.M. Intracerebral propagation of Alzheimer’s disease: Strengthening evidence of a herpes simplex virus etiology. Alzheimers Dement. 2013, 9, 169–175.
- Manuelidis, L. Infectious particles, stress, and induced prion amyloids: A unifying perspective. Virulence 2013, 4, 373–383.
- Köhler, C.; Maes, M.; Slyepchenko, A.; Berk, M.; Solmi, M.; Lanctôt, K.; Carvalho, A. The Gut-Brain Axis, Including the Microbiome, Leaky Gut and Bacterial Translocation: Mechanisms and Pathophysiological Role in Alzheimer’s Disease. Curr. Pharm. Des. 2016, 22, 6152–6166.
- Pistollato, F.; Sumalla Cano, S.; Elio, I.; Masias Vergara, M.; Giampieri, F.; Battino, M. Role of gut microbiota and nutrients in amyloid formation and pathogenesis of Alzheimer disease. Nutr. Rev. 2016, 74, 624–634.
- Li, C.-Q.; Zheng, Q.; Wang, Q.; Zeng, Q.-P. Biotic/Abiotic Stress-Driven Alzheimer’s Disease. Front. Cell. Neurosci. 2016, 10, 269.
- Chen, C.; Ahn, E.H.; Kang, S.S.; Liu, X.; Alam, A.; Ye, K. Gut dysbiosis contributes to amyloid pathology, associated with C/EBPβ/AEP signaling activation in Alzheimer’s disease mouse model. Sci. Adv. 2020, 6, eaba0466.
- Luca, M.; Di Mauro, M.; Di Mauro, M.; Luca, A. Gut Microbiota in Alzheimer’s Disease, Depression, and Type 2 Diabetes Mellitus: The Role of Oxidative Stress. Oxid. Med. Cell. Longev. 2019, 2019, 4730539.
- Akbari, E.; Asemi, Z.; Kakhaki, R.D.; Bahmani, F.; Kouchaki, E.; Tamtaji, O.R.; Hamidi, G.A.; Salami, M. Effect of Probiotic Supplementation on Cognitive Function and Metabolic Status in Alzheimer’s Disease: A Randomized, Double-Blind and Controlled Trial. Front. Aging Neurosci. 2016, 8, 256.
- Noble, J.M.; Scarmeas, N.; Celenti, R.S.; Elkind, M.S.V.; Wright, C.B.; Schupf, N.; Papapanou, P.N. Serum IgG Antibody Levels to Periodontal Microbiota Are Associated with Incident Alzheimer Disease. PLoS ONE 2014, 9, e114959.
- Xin, Y.; Diling, C.; Jian, Y.; Ting, L.; Guoyan, H.; Hualun, L.; Xiaocui, T.; Guoxiao, L.; Ou, S.; Chaoqun, Z.; et al. Effects of Oligosaccharides from Morinda officinalis on Gut Microbiota and Metabolome of APP/PS1 Transgenic Mice. Front. Neurol. 2018, 9, 412.
- Brandscheid, C.; Schuck, F.; Reinhardt, S.; Schäfer, K.-H.; Pietrzik, C.U.; Grimm, M.; Hartmann, T.; Schwiertz, A.; Endres, K. Altered Gut Microbiome Composition and Tryptic Activity of the 5xFAD Alzheimer’s Mouse Model. J. Alzheimers. Dis. 2017, 56, 775–788.
- Bonfili, L.; Cecarini, V.; Berardi, S.; Scarpona, S.; Suchodolski, J.S.; Nasuti, C.; Fiorini, D.; Boarelli, M.C.; Rossi, G.; Eleuteri, A.M. Microbiota modulation counteracts Alzheimer’s disease progression influencing neuronal proteolysis and gut hormones plasma levels. Sci. Rep. 2017, 7, 1–21.
- Du, X.T.; Wang, L.; Wang, Y.J.; Andreasen, M.; Zhan, D.W.; Feng, Y.; Li, M.; Zhao, M.; Otzen, D.; Xue, D.; et al. Aβ1-16 can aggregate and induce the production of reactive oxygen species, nitric oxide, and inflammatory cytokines. J. Alzheimer Dis. 2011, 27, 401–413.
- Bhatti, A.B.; Usman, M.; Ali, F.; Satti, S.A. Vitamin supplementation as an adjuvant treatment for Alzheimer’s disease. J. Clin. Diagn. Res. 2016, 10, OE07–OE11.
- Ono, K.; Yamada, M. Vitamin A and Alzheimer’s disease. Geriatr. Gerontol. Int. 2012, 12, 180–188.
- Sung, S.; Yao, Y.; Uryu, K.; Yang, H.; Lee, V.M.Y.; Trojanowski, J.Q.; Praticò, D. Early vitamin E supplementation in young but not aged mice reduces Abeta levels and amyloid deposition in a transgenic model of Alzheimer’s disease. FASEB J. 2004, 18, 323–325.
- Conte, V.; Uryu, K.; Fujimoto, S.; Yao, Y.; Rokach, J.; Longhi, L.; Trojanowski, J.Q.; Lee, V.M.Y.; McIntosh, T.K.; Praticò, D. Vitamin E reduces amyloidosis and improves cognitive function in Tg2576 mice following repetitive concussive brain injury. J. Neurochem. 2004, 90, 758–764.
- Lim, G.P.; Chu, T.; Yang, F.; Beech, W.; Frautschy, S.A.; Cole, G.M. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J. Neurosci. 2001, 21, 8370–8377.
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Curcumin Has Potent Anti-Amyloidogenic Effects for Alzheimer’s β-Amyloid Fibrils In Vitro. J. Neurosci. Res. 2004, 75, 742–750.
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901.
- Zhang, L.; Fiala, M.; Cashman, J.; Sayre, J.; Espinosa, A.; Mahanian, M.; Zaghi, J.; Badmaev, V.; Graves, M.C.; Bernard, G.; et al. Curcuminoids enhance amyloid-β uptake by macrophages of Alzheimer’s disease patients. J. Alzheimer Dis. 2006, 10, 1–7.
- Balogun, E.; Hoque, M.; Gong, P.; Killeen, E.; Green, C.J.; Foresti, R.; Alam, J.; Motterlini, R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem. J. 2003, 371, 887–895.
- Wruck, C.J.; Claussen, M.; Fuhrmann, G.; Römer, L.; Schulz, A.; Pufe, T.; Waetzig, V.; Peipp, M.; Herdegen, T.; Götz, M.E. Luteolin protects rat PC 12 and C6 cells against MPP+ induced toxicity via an ERK dependent Keapl-Nrf2-ARE pathway. J. Neural. Transm. Suppl. 2007, 72, 57–67.
- Matsubara, E.; Bryant-Thomas, T.; Quinto, J.P.; Henry, T.L.; Poeggeler, B.; Herbert, D.; Cruz-Sanchez, F.; Chyan, Y.J.; Smith, M.A.; Perry, G.; et al. Melatonin increases survival and inhibits oxidative and amyloid pathology in a transgenic model of Alzheimer’s disease. J. Neurochem. 2003, 85, 1101–1108.
- Ono, K.; Hirohata, M.; Yamada, M. Ferulic acid destabilizes preformed β-amyloid fibrils in vitro. Biochem. Biophys. Res. Commun. 2005, 336, 444–449.
- Hamaguchi, T.; Ono, K.; Murase, A.; Yamada, M. Phenolic compounds prevent Alzheimer’s pathology through different effects on the amyloid-β aggregation pathway. Am. J. Pathol. 2009, 175, 2557–2565.
- Cornejo, A.; Aguilar Sandoval, F.; Caballero, L.; Machuca, L.; Muñoz, P.; Caballero, J.; Perry, G.; Ardiles, A.; Areche, C.; Melo, F. Rosmarinic acid prevents fibrillization and diminishes vibrational modes associated to β sheet in tau protein linked to Alzheimer’s disease. J. Enzym. Inhib. Med. Chem. 2017, 32, 945–953.
- Rong, H.; Liang, Y.; Niu, Y. Rosmarinic acid attenuates β-amyloid-induced oxidative stress via Akt/GSK-3β/Fyn-mediated Nrf2 activation in PC12 cells. Free Radic. Biol. Med. 2018, 120, 114–123.
- Franco, R.; Casanovas, B.; Camps, J.; Navarro, G.; Martínez-Pinilla, E. Antixoxidant Supplements versus Health Benefits of Brief/Intermittent Exposure to Potentially Toxic Physical or Chemical Agents. Curr. Issues Mol. Biol. 2021, 43, 47.
- Cuttler, J.M. Application of Low Doses of Ionizing Radiation in Medical Therapies. Dose Response 2020, 18, 1559325819895739.


