Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Li, Y. Archaea in Pigs. Encyclopedia. Available online: https://encyclopedia.pub/entry/13185 (accessed on 07 February 2026).
Li Y. Archaea in Pigs. Encyclopedia. Available at: https://encyclopedia.pub/entry/13185. Accessed February 07, 2026.
Li, Ying. "Archaea in Pigs" Encyclopedia, https://encyclopedia.pub/entry/13185 (accessed February 07, 2026).
Li, Y. (2021, August 16). Archaea in Pigs. In Encyclopedia. https://encyclopedia.pub/entry/13185
Li, Ying. "Archaea in Pigs." Encyclopedia. Web. 16 August, 2021.
Copy Citation
Archaea is identified as the key link in the interaction between gut microbiota and host metabo-lism. Studies on human and mice have reported archaea, especially methanogenic archaea, makes an important impact on the energy harvesting capacity of the host by improving fermentation. But, in pigs, the metabolic potential of archaea at different production stages are still largely unknown.
swine
archaea
energy metabolism
CAZyme genes
pig
1. Introduction
Before Dr. Carl Woese and his colleagues separated archaea (named “Archaebacteria” at that time) from the bacteria domain based on phylogenetic analysis of ribosomal RNA sequences, archaea were considered a subgroup of bacteria [1]. To highlight the differences between archaea and bacteria, Dr. Carl Woese formally proposed the name of “Archaea” in his publication [2]. With the advancement of research technologies and tools, a large number of novel archaea and their characteristics have been discovered.
Archaea have highly diverse energy sources and unique metabolic characteristics and cell physiology, making it possible for them to live in various extreme environments, such as extreme temperature [3], high-salt [4], extreme alkaline [5], or acidic environments [6]. At the same time, archaea were also detected in various non-extreme ecosystems, including soil, ocean, lake water, and habitats associated with human and animal hosts, and were confirmed to make significant contributions to the ecological cycle by previous studies [7][8]. As a “young” group with unique natural capabilities and biological characteristics, archaea have attracted increasing research interest in the past decade.
Unlike bacteria, archaea are one of the least studied and least understood members of the human microbiota. One of the reasons is that archaea only account for a very small proportion of the microbiota. A recent study among East Asians living in South Korea showed that archaea were detected in 42.47% (381/897) and constituted 10.24 ± 4.58% of total bacteria and archaea among positive subjects [9]. However, the relatively small proportion of archaea does not mean they are not important. For example, some species of archaea have been reported to reduce Trimethylamine-N-oxide (TMAO), a harmful product associated with several diseases [10], through methanogenesis. In addition, methanogenic archaea can remove H2, the end product of bacterial fermentation, to increase the efficiency of fermentation and improve the energy harvesting capacity of the host [11]. Two studies performed on adults and children report that hydrogen-utilizing methanogenic archaea are co-enriched with H2-producing bacteria, Prevotella species, in the gut of obese humans [12]. Furthermore, human gut colonization by archaea has been associated with several other gastrointestinal and metabolic diseases, such as chronic constipation [13], inflammatory bowel disease [14], and colorectal cancer [15].
In pigs, the major ecological niches harboring archaea along the gastrointestinal tract (GIT) include the cecum, colon, and rectum, according to Gresse et al. [16]. The genus Methanobrevibacter is predominant with a relative abundance of 57–100% of the archaeal communities in pigs and sows [16][17][18][19]. Additionally, the archaeal composition in the swine GIT is dynamic, according to Su et al. [17] and Federici et al. [20], and diet-driven [21]. These exploratory studies provided important information on archaea in pigs, however, their sample sizes were small, and the composition and metabolic potential of archaea at different production stages in pigs are still largely unknown.
2. The Diversity of Archaea in Pigs
Significant differences in archaeal alpha diversity between different countries were observed. Pairwise comparisons of countries showed that archaeal diversity (Shannon Index) of Chinese pigs were significantly lower than that of the French (Figure 1A: Kruskal–Wallis test, p < 0.001) and Danish groups (p < 0.001). No differences in the swine archaeal alpha diversity were observed between French and Danish (p = 0.918) pigs. Beta diversity of the archaeal communities was visualized on a principal coordinate analysis (PCoA) plot based on the Bray–Curtis distance matrix. Chinese pigs had significantly different archaeal communities from Danish (ANOSIM, p < 0.001, R = 0.808) and French (ANOSIM, p < 0.001, R = 0.634) pigs as demonstrated on the PCoA plot (Figure 1B). However, the archaeal community structures of the Danish and French pigs could not be distinguished (Figure 1B; ANOSIM, p < 0.001, R = 0.109).
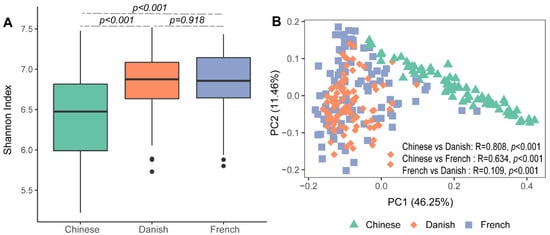
Figure 1. Swine archaeal alpha and beta diversities between three countries. Alpha diversity of archaeal communities in pigs was measured using the Shannon Index (A). Beta diversity was illustrated on a principal coordinate analysis (PCoA) plot based on Bray–Curtis distance (B).
3. The Archaeal Composition in Pigs
At the genus level, Methanobrevibacter, with an overall average relative abundance of 23.02%, was the most abundant archaeal genus (Figure 2), followed by Candidatus methanomethylophilus, which had a relative abundance of 10.52%. Of note, although Methanobrevibacter is considered the most dominant archaeal genus of pigs in general, it was ranked the second most abundant in Danish pigs. Additionally, Methanobrevibacter was significantly more abundant in Chinese pigs with the relative abundance of 44.94% than in both Danish (13.97%) and French pigs (15.41%), while Candidatus methanomethylophilus was decreased in Chinese pigs (0.98%) compared to Danish (15.71%) and French (12.59%) pigs.
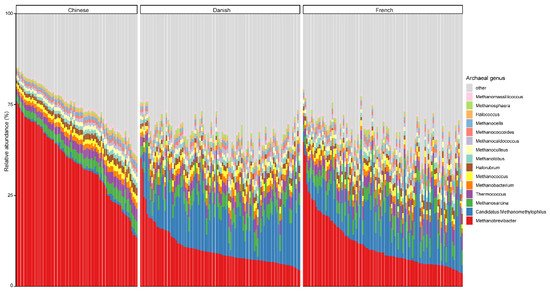
Figure 2. Relative abundance of the top 15 archaeal genera in swine fecal samples from different countries. Each column represents one pig, and each color represents one archaeal genus. The y-axis represents relative abundance ranging from 0 to 100%.
At the species level, the mean relative abundance of the top 15 species is shown in Figure 3. The gut microbiome of pigs is dominated by Natrialbaceae archaeon XQ INN 246 (mean relative abundance, 10.27%), followed by Candidatus methanomethylophilus alvus (9.57%), Methanobrevibacter gottschalkii (6.40%), and Methanobrevibacter smithii (3.42%). The most abundant species in different countries seem to be inconsistent, as well. The relative abundance of Candidatus methanomethylophilus alvus, Natrialbaceae archaeon XQ INN 246, and Methanobrevibacter gottschalkii were the highest in Danish, French, and Chinese pigs with an average relative abundance of 14.32, 11.67, and 16.28%, respectively.
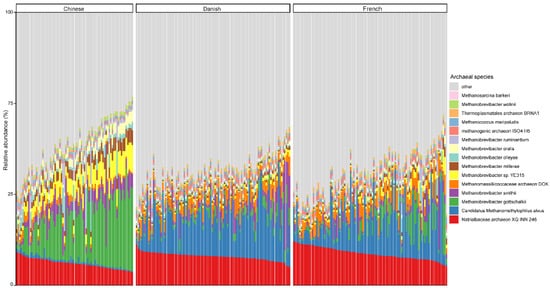
Figure 3. Relative abundance of the top 15 archaeal species in swine fecal samples from different countries. Each column represents one pig, and each color represents one archaeal species. The y-axis represents relative abundance ranging from 0 to 100%.
4. The metabolic Pathways of the Swine Archaea
To investigate the possible roles of gut archaea in swine, MetaCyc metabolic pathway profiles based on archaeal reads were investigated using HUMAnN3. A total of 77 MetaCyc pathways were observed, with 12 of these MetaCyc pathways present in more than 60% of fecal samples. Figure 4A shows the top six most abundant MetaCyc pathways. The most abundant MetaCyc pathway was involved in pyruvate fermentation to isobutanol (PWY-7111). Pathways of VALSYN-PWY and ILEUSYN-PWY ranked as second and third and were related to the biosynthesis of two essential amino acids, L-valine and L-isoleucine, respectively. Interestingly, these pathways, PWY-7111, VALSYN-PWY, and ILEUSYN-PWY, were also among the top 10 most abundant MetaCyc pathways in bacteria (Figure 4B). Furthermore, the pathway related to hydrogen consumption (METHANOGENESIS-PWY) was only observed in archaea and not in bacteria. In contrast, the pathways involved in hydrogen production (FERMENTATION-PWY and PWY4LZ-257) were only observed in bacteria and not in archaea (Figure 5).
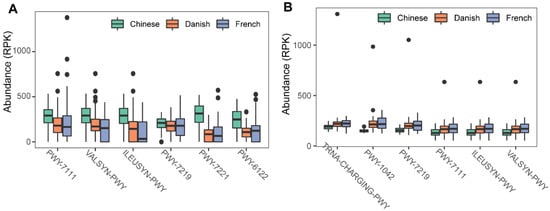
Figure 4. Summary of the predominant MetaCyc pathways of archaea and bacteria in pigs. The top six most abundant MetaCyc pathways of archaea (A) and bacteria (B) are displayed. In (A,B), the y-axis represents the number of reads mapped to reference genes involved in pathways. Data have been normalized to RPKM (reads per kilobase of transcript per million mapped reads).
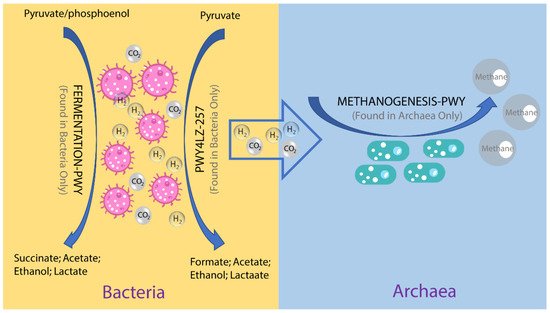
Figure 5. Overview of the hydrogen production pathways of bacteria and consumption pathway of archaea.
We next profiled the archaea-annotated CAZyme gene families in the swine gut. A total of 297 CAZyme gene families were identified from archaeal reads, out of which 99 were detected with a sample coverage of more than 60%. The three major CAZyme families were glycoside hydrolases (GH, 54/99), glycoside transferases (GT, 24/99), and carboxylesterase (CE, 11/99). The most abundant CAZyme category was AA3 (auxiliary activity family AA3), followed by GH43 and GT2 (Figure 6A). For comparison, we also investigated bacterial CAZyme profiles (Figure 6B) and found that six of the top 10 CAZyme families of archaea and bacteria were shared, including AA3, GT2, AA6, GT41, GT5, and GH13.
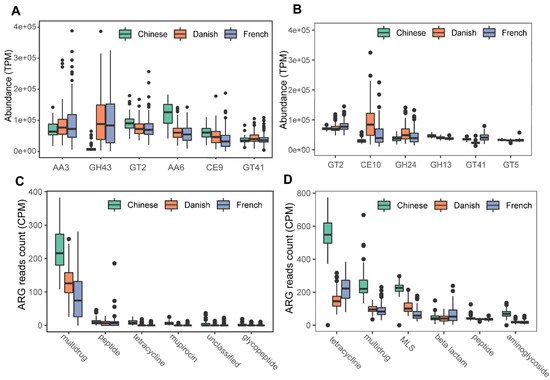
Figure 6. Summary of predominant CAZyme gene families (A,B) and antibiotic resistance genes (C,D) of archaea and bacteria in pigs. The top six CAZy gene families of archaea (A) and bacteria (B) were normalized by TPM (trans per million) using salmon software. ARG raw counts were identified using deeparg software. The ARG reads count of archaea (C) and bacteria (D) were normalized by CPM (counts per million).
Finally, we examined the ARGs in the swine gut archaea. In total, 13 classes of ARGs were detected from archaeal reads in all samples. On average, 0.035% of ARG hits were obtained from archaeal reads. The ARG class of multidrug resistance represented the vast majority (87.41%) of ARG hits; of the ARG class of multidrug resistance, the proportion of rpoB2 gene ranged from 87.18 to 100%. The other ARG classes with more than 1% relative abundance included peptide (5.07%), tetracycline (2.29%), mupirocin (1.69%), and unclassified (1.15%). The ARG type of multidrug class was significant enriched in Chinese pigs (Kruskal-Wallis test, p < 0.01), and the relative abundance of the multidrug class in Danish pigs is significantly higher than that in French pigs (Kruskal–Wallis test, p < 0.01). The boxplots of the top six ARG genes are shown in Figure 6C. In contrast, 24 ARGs were identified in bacterial reads, and 0.144% of bacterial reads were identified as ARG hits. The most abundant ARG class in bacteria was tetracycline, accounting for 40.59% of the total ARG hits (Figure 6D), while the class of multidrug ranked second with 19.39%, followed by MLS (17.73%), beta-lactam (7.46%), peptide (5.33%), and aminoglycoside (4.63%).
References
- Woese, C.R.; Fox, G.E. Phylogenetic structure of the prokaryotic domain: The primary kingdoms. Proc. Natl. Acad. Sci. USA 1977, 74, 5088–5090.
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579.
- Cavicchioli, R. Cold-adapted archaea. Nat. Rev. Microbiol. 2006, 4, 331–343.
- Andrei, A.-Ş.; Banciu, H.L.; Oren, A. Living with salt: Metabolic and phylogenetic diversity of archaea inhabiting saline ecosystems. FEMS Microbiol. Lett. 2012, 330, 1–9.
- Shen, J.p.; Zhang, L.m.; Zhu, Y.g.; Zhang, J.b.; He, J.z. Abundance and composition of ammonia-oxidizing bacteria and ammonia-oxidizing archaea communities of an alkaline sandy loam. Environ. Microbiol. 2008, 10, 1601–1611.
- Herbold, C.W.; Lehtovirta-Morley, L.E.; Jung, M.Y.; Jehmlich, N.; Hausmann, B.; Han, P.; Loy, A.; Pester, M.; Sayavedra-Soto, L.A.; Rhee, S.K. Ammonia-oxidising archaea living at low pH: Insights from comparative genomics. Environ. Microbiol. 2017, 19, 4939–4952.
- Moissl-Eichinger, C.; Pausan, M.; Taffner, J.; Berg, G.; Bang, C.; Schmitz, R.A. Archaea are interactive components of complex microbiomes. Trends Microbiol. 2018, 26, 70–85.
- Borrel, G.; Brugere, J.-F.; Gribaldo, S.; Schmitz, R.A.; Moissl-Eichinger, C. The host-associated archaeome. Nat. Rev. Microbiol. 2020, 18, 622–636.
- Kim, J.Y.; Whon, T.W.; Lim, M.Y.; Kim, Y.B.; Kim, N.; Kwon, M.-S.; Kim, J.; Lee, S.H.; Choi, H.-J.; Nam, I.-H. The human gut archaeome: Identification of diverse haloarchaea in Korean subjects. Microbiome 2020, 8, 114.
- Bain, M.A.; Faull, R.; Fornasini, G.; Milne, R.W.; Evans, A.M. Accumulation of trimethylamine and trimethylamine-N-oxide in end-stage renal disease patients undergoing haemodialysis. Nephrol. Dial. Transplant. 2006, 21, 1300–1304.
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027.
- Zhang, H.; DiBaise, J.K.; Zuccolo, A.; Kudrna, D.; Braidotti, M.; Yu, Y.; Parameswaran, P.; Crowell, M.D.; Wing, R.; Rittmann, B.E. Human gut microbiota in obesity and after gastric bypass. Proc. Natl. Acad. Sci. USA 2009, 106, 2365–2370.
- Pimentel, M.; Gunsalus, R.P.; Rao, S.S.; Zhang, H. Methanogens in human health and disease. Am. J. Gastroenterol. Suppl. 2012, 1, 28.
- Khelaifia, S.; Raoult, D. Haloferax massiliensis sp. nov., the first human-associated halophilic archaea. New Microbes New Infect. 2016, 12, 96.
- Coker, O.O.; Wu, W.K.K.; Wong, S.H.; Sung, J.J.; Yu, J. Altered gut archaea composition and interaction with bacteria are associated with colorectal cancer. Gastroenterology 2020, 159, 1459–1470.e1455.
- Gresse, R.; Chaucheyras Durand, F.; Dunière, L.; Blanquet-Diot, S.; Forano, E. Microbiota composition and functional profiling throughout the gastrointestinal tract of commercial weaning piglets. Microorganisms 2019, 7, 343.
- Su, Y.; Bian, G.; Zhu, Z.; Smidt, H.; Zhu, W. Early methanogenic colonisation in the faeces of Meishan and Yorkshire piglets as determined by pyrosequencing analysis. Archaea 2014, 2014, 547908.
- Luo, Y.-h.; Su, Y.; Wright, A.-D.G.; Zhang, L.-l.; Smidt, H.; Zhu, W.-Y. Lean breed Landrace pigs harbor fecal methanogens at higher diversity and density than obese breed Erhualian pigs. Archaea 2012, 2012, 605289.
- Mi, J.; Peng, H.; Wu, Y.; Wang, Y.; Liao, X. Diversity and community of methanogens in the large intestine of finishing pigs. BMC Microbiol. 2019, 19, 83.
- Federici, S.; Miragoli, F.; Pisacane, V.; Rebecchi, A.; Morelli, L.; Callegari, M.L. Archaeal microbiota population in piglet feces shifts in response to weaning: Methanobrevibacter smithii is replaced with Methanobrevibacter boviskoreani. FEMS Microbiol. Lett. 2015, 362, fnv064.
- Luo, Y.; Chen, H.; Yu, B.; He, J.; Zheng, P.; Mao, X.; Tian, G.; Yu, J.; Huang, Z.; Luo, J. Dietary pea fiber increases diversity of colonic methanogens of pigs with a shift from Methanobrevibacter to Methanomassiliicoccus-like genus and change in numbers of three hydrogenotrophs. BMC Microbiol. 2017, 17, 17.
More
Information
Subjects:
Agriculture, Dairy & Animal Science
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
813
Revisions:
2 times
(View History)
Update Date:
16 Aug 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No