 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mario Eduardo GUIDO | + 6200 word(s) | 6200 | 2021-08-02 16:30:25 | | | |
| 2 | Conner Chen | Meta information modification | 6200 | 2021-08-15 11:24:20 | | |
Video Upload Options
Gliomas are solid tumors of the central nervous system (CNS) that originated from different glial cells. The World Health Organization (WHO) classifies these tumors into four groups (I–IV) with increasing malignancy. Glioblastoma (GBM) is the most common and aggressive type of brain tumor classified as grade IV. GBMs are resistant to conventional therapies with poor prognosis after diagnosis even when the Stupp protocol that combines surgery and radiochemotherapy is applied. Nowadays, few novel therapeutic strategies have been used to improve GBM treatment, looking for higher efficiency and lower side effects, but with relatively modest results. The circadian timing system temporally organizes the physiology and behavior of most organisms and daily regulates several cellular processes in organs, tissues, and even in individual cells, including tumor cells.
1. GBM General Hallmarks
Gliomagenesis, as a multi-component process that promotes the development of gliomas, involves amplification and deletion or mutation of several genes, including the epidermal growth factor receptor (EGFR), tumor protein 53 (TP53), phosphate and tensin homolog (PTEN), and isocitrate dehydrogenase (IDH), among others [1]. These genes regulate distinct pathways known to be part of the core drivers of gliomagenesis, leading to aberrant signaling in proliferation, cell cycle regulation, senescence, and apoptosis [2][3].
One of the most studied hallmarks of human GBM is the amplification and genetic rearrangement of the gene that encodes for the tyrosine kinase receptor known as EGFR. This pathway can be activated either through overexpression of the receptor, amplification of the EGFR locus, and/or mutations in the receptor [4]. The most common and described mutation in GBM is the EGFRvIII, which corresponds to the loss of exons 2–7, resulting in a truncated extracellular domain with ligand-independent constitutive activity and consequently excessive cell proliferation. This mutation is associated with a bad prognosis and has not been observed in healthy tissues and secondary GBM subtypes [5]. Interestingly, GBM cells express either EGFR or EGFRvIII, although co-expression of both variants has also been reported in a small population of cells [6]. The TP53 gene encodes for a tumor suppressor protein that participates in cell cycle control, DNA damage response, cell death, and differentiation. Its mutation incidence is low in primary tumors (about 30%); however, 90% of secondary GBMs present mutations in this gene. Indeed, it has been proposed that TP53 mutation is an early event in secondary GBM [7] and is correlated with GBM progression by driving the activation of the mevalonate pathway since p53-mutant cells have shown an elevated activity of this pathway compared to wild type cells [8]. Other alterations related to this pathway include murine double minute 2 (MDM2) and MDM4 amplification, and CDKN2A-p14ARF deletion [2]. As MDM2 is a negative regulator of the TP53 gene, using inhibitors of MDM2 has shown promising results in GBM treatment [9]. PTEN gene is another commonly mutated tumor suppressor gene observed in most cancers, similar to TP53 [10]. It has a crucial role in inhibiting cell proliferation and regulating the migration and invasion of cells. PTEN is frequently inactivated in GBM, either by losing heterozygosity (LOH) of chromosome 10 or by mutation-induced constitutive activation of PI3K. The LOH of chromosome 10 is observed in almost 70% of GBM samples, predominantly in the primary subtype [1]. Amplification of platelet-derived growth factor receptor (PDGFR) is another genetic alteration observed in GBM tumors [11]. Lastly, IDH mutations are considered the most reliable indicator to differentiate primary from secondary GBM [12][13][14][15][16], primary GBM typically lacking IDH mutations [17]. The IDH gene encodes for isocitrate dehydrogenase, an enzyme that catalyzes the oxidative decarboxylation of isocitrate to 2-oxoglutarate within the Krebs cycle, whereas IDH mutants catalyze the production of the oncometabolite 2-hydroxyglutarate (2-HG). Importantly, patients treated with TMZ are associated with a favorable prognosis when they present IDH mutations since the synthesis of 2-HG interferes in the activation of DNA demethylation enzymes, yielding a hypermethylation status in tumor cells [18].
The O6-methylguanine-DNA methyltransferase (O6-MGMT) gene encodes for an enzyme that removes the methyl group from the guanine (position O6). The expression level of this protein is relevant to the treatment outcome when using TMZ since its expression is associated with a poor response. Consequently, the methylation level of its promoter is associated with a better response to TMZ treatment [1].
2. GBM Classification
The WHO classifies brain tumors into four groups (I–IV) of growing malignancy based on the morphological features of the tumor and their cells of origin [19]. Grades I and II include tumors with low proliferation potential, whereas grades III and IV tumors are high-grade gliomas characterized by high proliferation rates and aggressiveness [20]. GBM is classified as a grade IV high-speed growth tumor showing diffuse boundaries and is usually associated with a poor prognosis [7]. GBM is also divided into primary and secondary tumors, with primary GBM commonly diagnosed in the elderly without prior disease. Several alterations have been reported in primary GBM including LOH at 10q (70% of cases) [5][12][21] and 10p (50–70%) [5][22], amplification or mutation of EGFR (~35–45%) [5][12][21][22][23] mutation in TP53 (27–30%) [5][23], deletion of cyclin-dependent kinase inhibitor 2A/B (CDKN2A/B) (31%) [24], mutation or deletion of PTEN (25%) [5], promoter methylation of O6-MGMT (42%) [25], promoter mutation of telomerase reverse transcriptase (TERT) (72%) [21][23], mutation in glioma-associated oncogene homolog 1 (GLI1) (5–22%), deletion or mutation in phosphatidylinositol-4,5-bisphosphate 3-kinase A (PIK3CA) (~1%), MDM2 (7–12%) [21][22][24] and neurofibromatosis type1 (NF1) (11%), amplification of PDGFR (7%), and mutation in IDH1/2 (5%) [24].
On the other hand, secondary GBM develops from a low-grade glioma or an anaplastic astrocytoma, affects younger persons, and shows a better prognosis after diagnosis. These tumors are much less common, showing genetic alterations that include mutation in IDH1/2 (73–85%) [26], TP53 (65–81%), ATRX (~65–71%) [5][23], and PTEN (<5%) [5] genes, promoter methylation of MGMT (79%) [25], loss of chromosome 19q (~50%) and 10q (63%) [5], p16INK4a deletion (19%) and EGFR amplification (8%) [12]. Mutant IDH1 is considered a metabolic marker of secondary GBM because of its ubiquitous expression in lower-grade gliomas that eventually progress to GBM. Besides the differences described above regarding genetic profiles, primary and secondary GBMs are histologically indistinguishable, and the most reliable indicator to differentiate them are mutations in the IDH1 gene [16]. A summary of the most frequent genetic alterations in primary and secondary GBMs is presented in Table 1.
Table 1. Genetic alterations of primary and secondary Glioblastoma (GBM).
| Primary GBM [5][23][24][25][26] |
Secondary GBM [5][12][25][26] |
|---|---|
| LOH chromosome 10q (70%) | IDH1/2 mutation (73–85%) |
| LOH chromosome 10p (50–70%) | TP53 mutation (65–81%) |
| EGFR amplification or mutation (35–45%) | ATRX mutation (65–71%) |
| TP53 mutation (27–30%) | LOH chromosome 10q (63%) |
| PTEN mutation (25%) | LOH chromosome 19q (~50%) |
| O6-MGMT promoter methylation (42%) | MGMT promoter methylation (79%) |
| TERT promoter mutation (72%) | p16INK4a deletion (~19%) |
| PDGFR amplification (~7%) | EGFR amplification (8%) |
| MDM2 mutation (7–12%) | PTEN mutation (<5%) |
| NF1 mutation/deletion (11%) | |
| GLI1 mutation (5–22%) | |
| IDH1/2 mutation (5%) | |
| PIK3CA mutation (1%) |
Summary of reported genetic alterations observed in primary and secondary GBM.
In 2016, GBM classification was updated, considering the specific molecular and genetic profiles observed in the different tumors [3][27]. Based on this, GBM was classified into four subtypes: proneural, neural, classic, and mesenchymal [7][1][28][29][30]. The proneural group constitutes the most frequent secondary GBM and has histological features most consistent with oligodendrocytes. This subtype, typically found in younger patients, is associated with the best prognosis after treatment. The most frequent genetic alterations observed in this subtype of GBM are mutations in PDGFRA, IDH1, TP53, and PIK3C genes. The neural profile is characterized by TP53 mutation, EGFR amplification, and CDKN2A deletion. The histology that describes this subtype is consistent with a combination of oligodendroglial, astrocytic, and neuronal features. Also, an important composition of genes involved in nervous system development and function (NEFL, GABRA1, SYT1, and SLC12A5) and a greater degree of neuronal marker expression was observed in the neural subtype. The classic or proliferative subtype is associated with EGFR amplification (97%), LOH of chromosome 10, chromosome 7 amplification, and CDKN2A-p16INK4a deletion (94%) and demonstrated features more consistent with astrocytes. The mesenchymal subtype of GBM is associated with a worse prognosis and evidence of a greater degree of necrosis and inflammatory components. This profile is characterized by overexpression of mesenchymal and astrocytic markers, lower expression of the tumor suppressor NF1, altered PTEN, TP53, CDKN2A, Akt genes, and the presence of mesenchymal markers (MET, CHI3L1, CD44, and MERTK). The classical and mesenchymal subtypes are associated with more aggressive high-grade gliomas, the worst prognosis compared to other profiles, and a slightly better response to aggressive therapies [1][27][28][30][31]. Table 2 summarizes the most important features of the four subtypes described above.
Table 2. Features of neural, proneural, classical, and mesenchymal GBM subtypes.
| GBM Subtype | Molecular and Genetic Profile | Median Survival (Months) |
|---|---|---|
| Proneural [7][1][27][28][30] |
|
11.3 (9.3–14.7) |
| Neural [1][27][30] |
|
13.1 (9.8–18) |
| Classical [1][27][28][30] |
|
12.2 (11.08–18) |
| Mesenchymal [1][27][28][30] |
|
11.8 (9.57–15.4) |
Summary of features described in the literature for neural, proneural, classical, and mesenchymal GBM subtypes.
In addition to the molecular and genetic features, the different subtypes of GBM have also been associated with the distinct localization of the tumors in the brain. Regarding the anatomical localization of the different subtypes, it was reported that tumors belonging to proneural and neural subtypes are found in the subventricular zone (SVZ) and showed a more rapid progression and poorer response to treatment compared to those localized outside of the SVZ of the brain. On the contrary, classical and mesenchymal GBMs are localized diffusely and away from the SVZ [32]. Interestingly, a recent transcriptome analysis revealed only three subtypes of GBM, presenting strongly enriched mRNAs associated with classical, proneural, and mesenchymal subtypes. This observation suggests that the neural subtype may represent a contamination of the original samples with non-tumor cells [33]. Regarding the response of the different GBM subtypes to treatment, it was observed that an aggressive therapeutic approach significantly reduces the mortality of patients with tumors belonging to the classical and mesenchymal subtypes. On the contrary, patients diagnosed with the neural subtype showed a slight response to this treatment, and non-significant changes were observed in the proneural patient cohort [27].
3. Circadian Rhythms
The circadian clocks (from Latin circa: near/dies: day) temporarily regulate cell-autonomous oscillations with a 24-h periodicity of a large array of biological processes and behaviors such as sleep/wake cycles, feeding/fasting control, metabolism, hormone secretion, and immune function [34]. The evolutionarily conserved circadian mechanism in the diverse species studied (reviewed in [35]) is made up of central and peripheral oscillators distributed in organs, tissues, and even in individual cells [34]. The suprachiasmatic nuclei (SCN) in the anterior hypothalamus harbors the master circadian clock, which is synchronized by external cues, or Zeitgebers (timer-givers) such as light or temperature, among others to anticipate and adapt the circadian timekeeping system to the environment [36][37][38].
Light and the environmental illumination conditions are the strongest synchronizers of the SCN through the projections from the retina [36][39]. The master clock can coordinate circadian outputs to synchronize peripheral clocks (e.g., liver, kidney, skin, intestine, lung, pancreas, ovary, and heart) in a tissue-specific manner through the autonomic nervous and the neuroendocrine systems [37][40]. Therefore, the central clock and peripheral oscillators drive the rhythmic expression of genes to couple physiological and behavioral processes to periodic environmental changes. However, modern life characterized by increased night-time activities with prolonged artificial lighting such as rotating shift work, hypercaloric diets, shortened sleep hours, and jet lag alters endogenous homeostasis with external cues. Consequently, this misalignment characterized by loss of the correct coordination between elements of the circadian system is considered a contributing factor to the development of metabolic syndrome, inflammatory disorders, and higher cancer risk [41][42][43][44].
4. The Molecular Clock
In 2017, the Nobel Prize in Physiology and Medicine was awarded to J.C. Hall, M. Rosbash, and M.W. Young for describing the molecular clock mechanism that underlies the circadian rhythms using fruit flies as a model organism. They showed that a gene named Period encodes for a protein whose expression was regulated by a negative feedback loop [45]. Later, other proteins of the circadian machinery were identified and extended to other species to elucidate the molecular clockwork mechanism in the cell. In mammals, the molecular clock comprises the so-called transcriptional/translational feedback loops (TTFL) [35][46] including a core set of clock genes that encodes positive and negative regulators. The primary loop involves the positive elements Bmal1 (aryl hydrocarbon receptor nuclear translocator-like, Arntl) and Clock (circadian locomotor output cycles kaput) and its paralogue neuronal PAS domain protein 2, Npas2, and the negative components Per1/2 (Period) and Cry1/2 (Cryptochrome) genes. During the day, the CLOCK:BMAL1 heterodimer recognizes the E-box sequence in Per and Cry promoters, increasing the levels of these transcripts. Then, PER and CRY proteins form a repressor complex that translocates to the nucleus and represses the CLOCK:BMAL1 activity inhibiting their expression. During the night, the repressor complex of PER and CRY is degraded, allowing CLOCK:BMAL1 to activate a new cycle of transcription and translation of approximately 24 h [47].
Furthermore, post-translational modifications of PER and CRY proteins such as phosphorylation-dependent ubiquitination and proteasomal degradation occur in a circadian manner and regulate the subcellular localization and/or half-life of proteins contributing to the progression and beginning of a new 24-h cycle [48][49][50][51][52]. In the secondary loop, the CLOCK:BMAL1 heterodimer activates the transcription of Rev-erbα/β genes, which belong to the orphan retinoic acid receptors family. In turn, REV-ERB proteins compete with ROR receptors for binding to ROR-response elements (RORE) sequences in the Bmal1 promoter to repress or activate their transcription, respectively [53][54]. Also, the CLOCK:BMAL1 heterodimer regulates the expression of a set of genes known as clock-controlled genes through E-box sites in their promoter regions. In this way, the circadian clock exerts its control in molecular, biochemical, and physiologic processes, including cell cycle, proliferation, metabolism, senescence, and DNA repair, among others [55][56][57][58]. In addition, at the SCN, fluid communication between astrocytes and neurons was observed to coordinate and maintain circadian oscillations [59].
5. Circadian Disruption and Its Implication in Cancer Biology
As postulated by Hanahan and Weinberg (2011), tumor cells share common features known as hallmarks of cancer that characterize how cancerous cells disrupt cellular homeostasis promoting tumor growth. These hallmarks include sustaining signaling promoting cellular proliferation, replicative immortality, the capability of evading cell death mechanisms, the capacity to avoid growth suppressors, the ability to trigger blood vessel formation (angiogenesis), the capacity of metastasis, the deregulation of cellular energetics, and the capability to evade the anti-tumoral immunological response. The features mentioned above are associated with crucial genomic instability and inflammation, contributing to tumor development [60]. Several studies in the literature suggest tight crosstalk between the circadian clock function with tumorigenesis and cancer progression in different tumor models. It was evidenced that clock and clock-controlled genes regulate several pathways involved in cellular proliferation and growth under physiological conditions and that, when altered, may promote some of the hallmarks mentioned above of cancer, strongly suggesting that tumor cells can hijack the endogenous clock functioning to assure unrestricted proliferation, enhance the metabolism to supply their high energetics demands, and adapt and modify the microenvironment to promote tumor growth [61][62].
The molecular clock can positively or negatively modulate the different cell cycle phases. Moreover, several regulators implicated in cell cycle checkpoints show daily expression patterns [63][64][65][66]. Sustaining cell signaling is considered another hallmark of cancer, and several studies suggest its connection with the circadian clock, showing that proteins related to proliferation pathways exhibited circadian patterns of expression [63][67]. In this respect, Myc oncogenic activation is also observed when deregulation of sympathetic nervous system modulation of peripheral tissues occurs [68]. Also, the circadian expression, stability, and activity of p53, one of the most studied tumor suppressors, is modulated by BMAL1 and PER2 [69][70][71][72][73]. Lastly, the RAS/MAPK pathway was associated with alterations in the circadian clock, in which anomalous RAS activation impairs CLOCK:BMAL1 activity and up-regulates Ink4a/Arf [74][75].
Regarding tumor metabolism, cancer cells have high energetic demands to sustain exacerbated proliferation. Circadian disruption has been associated with changes in the cellular metabolic program, modulating glucose utilization, amino acid uptake, lipogenesis, glycerophospholipid metabolism, and β-oxidation [43][76][77][78]. This metabolic rearrangement is known as the Warburg effect, in which metabolism mainly occurs through glycolysis, as opposed to mitochondrial oxidative phosphorylation in normal cells. Also, this phenomenon is associated with a reduction in the tricarboxylic acid (TCA) cycle activity, an increase in the synthesis of fatty acids, and an enhanced NADPH formation. Remarkably, the circadian clock regulates NADPH levels, a critical anabolic intermediate that plays a crucial role in cancer development [79]; it also regulates the expression of several genes involved in the transport and metabolism of glucose [80][81][82].
Since the circadian clock plays an essential role in immune system regulation, alterations in clock function have been associated with aberrant inflammation, evasion of immunological surveillance, and immune cell functionality changes leading to cancer progression [83][84]. Lastly, cell death and DNA damage response are other mechanisms involved in the recognized hallmarks of cancer associated with the circadian clock deregulation [62][85][86][87][88][89][90][91][92][93] (Figure 1).
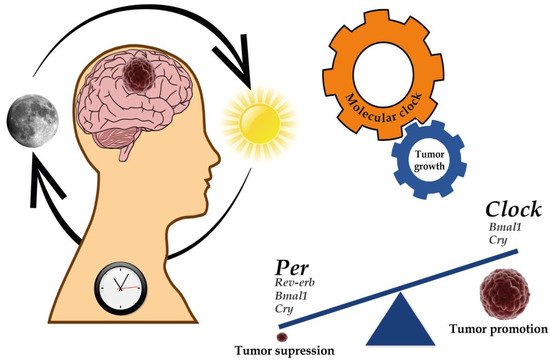
Figure 1. Implications of the circadian clock on cancer development and progression. Several genes of the molecular clock have been associated with both the regulation of GBM genesis and progression as well as tumor suppression. For further details, please see Table 3.
Table 3. Evidence of dysregulated clock gene expression in GBM.
| Gene | Evidence of Gene Deregulation Associated with Gliomagenesis—Evidence of Potentiality for Therapeutic Targeting |
|---|---|
| Clock | [94][95][96][97][98][99][100][101][102] |
| Bmal1 | [103][104][76][94][97][105][106][107] |
| Per1 | [103][76][96][100][102][108][109][110][111] |
| Per2 | [96][102][108][110][111][112][113] |
| Per3 | [96][100][102][111] |
| Cry1 | [94][107][100][102][111][114] |
| Cry2 | [96][107][100][111][114][115] |
| Npas2 | [100][102] |
| Rev-erb | [94][116][96][97][117][118] |
| RORα | [96] |
| RORβ | [96] |
| Timeless | [102][119] |
Summary of references available in the literature regarding the implication of circadian clock genes in the biology of GBM.
6. The Positive Arm of the Molecular Clock
6.1. Bmal1 Gene
BMAL1 and its binding partner CLOCK recognize the E-box motif in the promoter of clock and clock-controlled genes, activating their transcription. Since BMAL1 was shown to regulate several critical cellular processes such as cell cycle progression, lipid, and glucose metabolism, redox state, and stress response [120][121][122], this highlights the putative crosstalk between the molecular clock and cancer development and progression. However, controversial results about the role of BMAL1 suggest that its function is tissue- and cancer-specific. In gliomas, either upregulation or downregulation of BMAL1 expression has relevant repercussions on their biology. Upregulation in the expression of BMAL1 was reported in the analysis of the TCGA database in high-grade glioma patients [94].
Additionally, BMAL1 knockdown impaired proliferation of patient-derived GSCs in culture, inducing cell cycle arrest and apoptosis as well as extending the life span and inhibiting tumor growth in a murine model [94][97]. Interestingly, targeting BMAL1 unaltered the normal neural stem cell proliferation, suggesting a critical role for this circadian regulator on GSCs growth and survival. The mechanism proposed by Dong et al. (2019) suggests that GSCs reprogram their metabolism through the molecular clock and epigenetic modifications since BMAL1 preferentially binds to the promoter region of genes involved in critical metabolic pathways such as those of glycolysis and TCA cycle [94].
It was also proposed that BMAL1 may act as a tumor suppressor in GBM cell growth. For instance, Jung et al. (2013) reported that BMAL1 overexpression impairs glioma invasiveness by blocking the PI3K/AKT/matrix metalloproteinase-2 signaling pathway [105]. In concordance with this, BMAL1 overexpression significantly decreases U-87MG cell viability (a cellular model of GBM) and Cyclin B1 levels, which play a critical role in the G2/M transition cell cycle. Also, the expression of pro-apoptotic proteins was increased while the anti-apoptotic protein BCL-2 level decreased, suggesting that BMAL1 may operate as a tumor-suppressor in U-87MG cell cultures. Glioma migration and invasion were also reduced after ectopic expression of BMAL1, leading to downregulation of p-AKT and MMP-9 signaling pathways [106]. Similar to the observations described above, results obtained recently by Wagner et al. (2021) show that the downregulation of Bmal1 expression is associated with a more aggressive form of the tumor. In this study, a cell line isolated from a malignant peripheral nerve sheath tumor generated in NPcis mice (an animal model for the human neurofibromatosis type I) was used as a glioma model after being injected into C57BL/6 animals and tumor growth evaluated. The results showed that, after the knockdown of Bmal1 using CRISPR/Cas9, tumors grew faster than those from control cells [103]. Suliman Khan et al. (2019) identified oncogenes and tumor suppressor genes that show significant variations in their expression in brain tissues from animals exposed to a CJL protocol. Interestingly, this study uses Bmal1−/− animals and suggests that expression of some of these genes is associated with the clock, highlighting the link between circadian disruption by CJL and the risk of glioma development [107]. More studies are needed to fully comprehend the biological importance of the circadian transcriptional regulator BMAL1 in the genesis and progression of brain tumors.
6.2. Clock Gene
As it was reported for its binding partner BMAL1, TCGA database analysis revealed that the Clock gene, located at 4q12 chromosomal region, is amplified in ~5% of GBM patients [120][105], and high-grade gliomas exhibited an increased expression of CLOCK compared to low-grade glioma or non-tumor cells [95][98][99][100]. An exploratory study carried out by Madden et al. found that CLOCK was overexpressed in tumors and that a single nucleotide polymorphism (rs7698022) present in the Clock gene was correlated with mortality in high-grade glioma patients [100]. A report in the literature indicates that CLOCK explicitly modulates the proliferation and cell death after irradiation in U-87MG cells. After Clock silencing, a reduction in proliferation and induction of apoptosis was observed in glioma cells. This phenomenon was associated with a downregulation of c-Myc and Cyclin B1 and upregulation of p53 related genes. These results highlight the anti-apoptotic modulation of CLOCK in glioma cells [101]. In human GSCs, CLOCK was proposed as a critical regulator of metabolism required for optimal GSC growth and survival since CLOCK depletion impaired GSCs self-renewal, reduced enzyme expression involved in glycolysis and TCA, and triggered cell cycle arrest and apoptosis [94][97]. These results agree with the findings described above by Dong et al. (2019) that evidence the crucial role of BMAL1 and CLOCK in tumor metabolism and stemness maintenance [94].
Additionally, Chen et al. (2020) suggested that CLOCK is implicated in the modulation of immune-suppressive microglia infiltration into the tumor microenvironment, seemingly by regulating the expression of the chemokine OLFML3. Interestingly, in the results obtained using an in vivo model of GBM, it was observed that downregulating the expression of CLOCK or OLFML3 shows an extension in the lifespan of mice compared to the control group [97]. More evidence suggests that CLOCK has a tumor-promoting function in gliomas. Li et al. (2013) showed in a fascinating study that high expression of CLOCK observed in high-grade gliomas tissues and GBM cell lines is associated with an attenuated miR-124 expression. This miRNA specifically targets the 3′UTR of Clock mRNA, and it was previously reported to impair cell proliferation and migration of tumor cells. Remarkably, CLOCK might promote the proliferation and migration of glioma cells through the NF-kB signaling pathway [95]. By contrast, the report from Wang et al. (2021) shows that CLOCK is downregulated in GBM samples [102].
Overall, the results discussed above suggest that CLOCK may promote tumor proliferation in different glioma models and play a critical role as a regulator of tumor metabolism. Considering this evidence, targeting the circadian clock by “adjusting the CLOCK” could be a promising strategy for GBM treatment, especially to impair GSC growth.
7. The Negative Arm of the Molecular Clock
7.1. Period 1 Gene
Per1 encodes for PER1 protein, a negative element of the circadian transcriptional machinery. Early studies from Wang’s laboratory showed that PER1 expression is lower in high-grade gliomas than in the surrounding non-tumor tissues. This study suggested that the deregulation in PER1 expression allows glioma cells to proliferate and survive, as this was related to a disruption of the clock function [108]. Similar results recently showed a reduction in PER1 expression in high-grade gliomas [102]. In agreement with these observations, tumors generated by injection of cells isolated from a malignant peripheral nerve sheath tumor exhibited lower Per1 mRNA levels than normal tissue [103]. One possible explanation linking the low levels of Per1 mRNA and protein with a higher tumor malignancy may be associated with the opposite relationship found between its expression and the phospholipid biosynthesis required for the genesis of the new membranes and other essential processes during cell growth and proliferation, as was observed in a non-malignant fibroblast cell line [77].
On the other hand, the analysis performed by Madden et al. (2014) found overexpression of PER1 and identified a PER1 variant (rs2289591) associated with glioma risk and, similar to the CLOCK variant described above, it was associated with mortality in high-grade glioma patients [100]. Interestingly, Per1 expression was related to the radiosensitivity of gliomas in culture; Per1 downregulation attenuated U343 glioma cell radiosensitivity, decreasing the apoptosis of irradiated tumor cells. Since PER1 knockdown decreased the levels of CHK2 and p53 proteins, critical checkpoints in DNA damage, the authors suggest that PER1, as a tumor suppressor gene, modulates the p53 pathway and, in consequence, influences p53 levels with a direct effect on apoptosis promotion and proliferation inhibition [109]. Similarly, high expression of Per1 correlated with increased radiosensitivity in gliomas cells in a rat model, while this phenomenon was not evidenced in non-tumor tissues. This study observed that Per1 levels show a circadian expression pattern in both normal and tumor tissues. However, glioma tissues evidenced a 12-h periodicity on Per1 expression while normal tissues displayed oscillations with a period close to 24 h. Like the previously described report, the author highlights the tumor suppressor role of PER1 in gliomas, showing that its expression is related to cell cycle arrest and enhanced x-ray sensitivity [110]. Also, findings from our laboratory demonstrated a 28 and 16-h rhythmicity on Per1 mRNA levels in arrested and proliferative T98G cultured cells, respectively [76]. A recent report by Gao et al. (2021) shows that the IDH1 mutation (R132H) is associated with a reduction in GBM cell proliferation as well as with the modification in clock gene levels, including a decrease in the expression of PER1 [111]. Besides the downregulation of Per1 levels observed in gliomas, these results suggest that tumor cells may display aberrant oscillations on Per1 expression, influencing cell proliferation and tumor survival.
7.2. Period 2 Gene
PER2 protein expression in gliomas has also been reported to be disturbed in comparison with normal brain tissues. Early results showed that PER2 expression was significantly lower compared to non-glioma cells, bringing out differences in the expression of clock genes between normal and malignant brain tissues [108]. Later and in concordance with the previous report, Wang et al. (2014) analyzed the expression of PER2 in glioma samples by immunohistochemistry and found a significant reduction in PER2 expression associated with high-grade gliomas and higher expression of EGFR and PCNA. Additionally, the authors proposed that promoter methylation or cell signaling pathway disruption may influence PER2 expression in tumor tissues [112]. In the same line, PER2 was found to be downregulated in samples from the TCGA database [99], and deregulation in PER2 tumor expression was associated with higher mortality in the cohort of glioma patients [96]. Similar to that observed for PER1, PER2 expression was associated with effectiveness in radiotherapy, again supporting the hypothesis that both genes are tumor suppressors [110].
A crucial role of PER2 in gliomagenesis was recently informed. Per2 mRNA and protein levels were reported to be downregulated in GSCs, and its overexpression impairs its proliferation through the cell cycle, arresting them in G0/G1 phase. The authors suggest that since PER2 targets the Wnt/β-catenin signaling pathway in GSCs, the downregulation of critical proteins involved in the invasiveness and stemness of GSCs, such as Wnt7b, β-catenin, MMP2, MMP9, and c-Myc, may explain the tumor suppressive role of PER2 in gliomas [113]. The IDH1 R132H mutation was also associated with a decrease in protein levels for PER2 [111].
7.3. Period 3 Gene
PER3 expression decrease in gliomas has been observed and related to higher mortality [96][100]. Wang et al. (2021) showed that the analysis of TCGA samples indicates a reduced PER3 expression in GBM samples [102]. Moreover, IDH1 R132H mutation was associated with a reduction in PER3 expression level [111]. The above observations suggest that similar to PER1 and PER2, PER3 could be crucial to gliomagenesis, acting as a tumor suppressor gene. Nevertheless, the role of PER3 in gliomagenesis and progression needs to be further investigated.
7.4. Cryptochrome 1 Gene
The circadian proteins CRY and PER form a repressor complex that inhibits their transcription, and that of other clock-controlled genes once translocated to the nucleus and represses the CLOCK:BMAL1 heterodimer transcriptional activity. Therefore, CRY as well as PER proteins are critical factors in the maintenance of cellular circadian homeostasis. A study of 69 sample patients evidenced a downregulated expression of Cry1 in glioma tissues compared with non-tumor cells [114]. Conversely, TCGA database analysis reported higher levels of Cry1 in GBM patients than in normal brains [100][102]. In U-87MG cell cultures, mutations in the IDH1 gene significantly correlated with a downregulated Cry1 expression compared to control cells. This study model proposed that IDH1 mutation affects glioma proliferation by altering clock gene expression through the TGF-ß/Smad signaling pathway [111]. Also, the role of Cry1 on glioma biology has been evidenced in experimental models of Cry1/2 double knockout mice subjected to CJL conditions. These results suggested a link between clock genes and glioma-related genes as well as the implication of lighting conditions in carcinogenesis [107]. Lastly, recent research proposed the pharmacological modulation of the circadian clock as a novel strategy for GBM treatment. KL001 is a synthetic agonist that stabilizes CRY protein levels, preventing their degradation. Dong´s laboratory showed that KL001 treatment impaired GSC proliferation and decreased stem cell markers expression [94].
7.5. Cryptochrome 2 Gene
In glioma tissues, expression of Cry2 was attenuated compared to healthy samples [102][114] and correlated with higher mortality [96]. However, findings of irradiated glioma cells in a rat model showed a correlation between the increased expression of Cry2 and increased cell proliferation and decreased apoptosis. As mentioned above, for Per1 expression, disturbances on rhythmic expression of Cry2 were observed in glioma tissues with a period of 8 h compared to 24-h periodicity displayed by normal brain samples suggesting that an altered rhythmic expression of Cry2 influences sensitivity to irradiation on gliomas cells [115]. Similar to results observed on Cry1 expression, IDH1 mutated U-87MG cells showed lower levels of Cry2 than wild-type cells with implications in tumor proliferation [111]. In view of the findings described, it can be inferred that the expression of the negative circadian regulator Cry2 is altered in gliomas tissues. Nevertheless, further investigation is needed to elucidate the key role of Cry2 in GBM development.
7.6. Rev-Erb Genes
NR1D1 and NR1D2 genes encode for the nuclear receptors known as REV-ERBα and REV-ERBβ, respectively. These nuclear receptors play critical functions in circadian rhythms, lipid and glucose metabolism, tumorigenesis, and inflammation and have been proposed to act as the molecular clock components linking the circadian clock with the cellular metabolism [123][124][125][126][127]. In glioma tissues, REV-ERBβ levels are lower compared to non-glioma tissues [96]. Conversely, Yu et al., (2018) reported a high expression of REV-ERBβ in GBM tissues and cell lines which was not observed in primary human astrocytes. Results from this laboratory suggested that NR1D2 is involved in the migration and invasion of glioma cells through the receptor tyrosine kinase AXL [116]. Synthetic agonists of the nuclear receptors REV-ERBs (SR9009 and SR9011) have been selectively lethal in different cancer cell lines, including GBM. For instance, T98G cells showed the highest response to SR9009 treatment in a time window from 18 to 30 h post synchronization with dexamethasone [117]. Since SR9009 can cross the BBB [124], REV-ERBs agonists have emerged as an interesting approach to GBM treatment. In vivo experiments showed that SR9009 treatment impaired glioma growth and improved survival in mice [97][118]. Remarkably, SR9009 efficacy to reduce tumor growth was similar to that observed with the current standard of care treatment, TMZ. Sulli et al. (2018) proposed that the pharmacological modulation of the circadian clock by REV-ERBs agonists impairs tumor proliferation, inhibiting de novo lipogenesis and autophagy, which are well-known hallmarks of tumor cells [118]. Further evidence show alterations in tumor metabolism after SR9009 treatment, as is the case of T98G cells that increase the average size of lipid droplets [117] and GSCs, which reduce the expression of genes involved in glycolysis, the TCA cycle, and lipid metabolism [94]. Considering that REV-ERBs inhibit Bmal1 expression, agonists of these nuclear receptors could be regarded as an exciting novel approach to target Bmal1, which has been shown to have tumor-promoting features in GBM models as discussed above.
7.7. Other Clock Pathways Related Genes
Other genes associated with the molecular clock were found to correlate with gliomagenesis and progression. NPAS2 is a protein coded by the Npas2 gene that heterodimerizes with BMAL1, and its expression in gliomas was associated with patients having poor outcomes and high mortality [100]. RORα and RORβ, which modulate the transcription of Bmal1, were observed to be downregulated in gliomas, and this expression profile was prognostic in a cohort of glioma-diagnosed patients suggesting that its function is associated with tumor genesis and progression [96]. TIMELESS is a protein belonging to the clock machinery, its activity regulates Clock, Per, and Cry gene expression and interacts with S-phase checkpoint proteins, having a crucial role in modulating the cell cycle. Recent work from Wang and Chen (2018) evidenced that TIMELESS is overexpressed in high-grade gliomas compared to low-grade glioma and non-pathological tissues. The authors suggested that this imbalance in the expression of the Timeless gene results in the abnormal progression of circadian rhythms and gliomagenesis promotion [119]. Similar to the former study, higher TIMELESS expression was observed in GBM compared to low-grade gliomas. Moreover, silencing of TIMELESS by siRNA leads to cell cycle arrest in G0 phase and cell proliferation impairment, again showing the importance of TIMELESS on GBM promotion by modulation of cell cycle and proliferation [102].
8. Chronotherapy as a Promising Strategy for GBM Treatment
Based on recent findings discussed in this review, the evidence accumulated to date clearly shows that the biological timekeeping system is intricately connected with cancer development and progression. The above-discussed literature highlights that more profound knowledge regarding the circadian modulation on cancer biology could either improve tumor treatment or develop new therapeutic strategies. Besides the promising agonists of the clock proteins that show antitumor activity on brain tumors, chronotherapy is a growing field of research that aims to improve the efficacy of current GBM treatment. Chronotherapy is defined as the drug delivery schedule based on patients ‘circadian rhythms, giving the drug administration timing an important role in therapy. This approach aims to determine the optimal time of the day to perform the treatment and improve outcomes with the most effective drug concentrations (not necessarily the highest dose used), and reduced drug toxicity and side effects.Early evidence in this area showed that the highest response to TMZ treatment occurs near the peak of Bmal1-luc expression in a murine cellular model of GBM in culture. Moreover, phosphorylation of the histone H2AX and activation of apoptosis after TMZ treatment displayed a circadian pattern that correlates with that observed for Bmal1 expression. Remarkably, the caspase activity oscillation induced by the DNA alkylator is abolished in Bmal1 knockout cells by CRISPR/Cas9 technology compared to control cells, suggesting a mechanism dependent on the Bmal1 clock gene expression [22]. In the same way, GBM T98G cells exhibited a significant temporal susceptibility response to the proteasome inhibitor Bortezomib which is used in advanced stages of GBM treatment. Bortezomib-treated cells display the highest susceptibility in a time window ranging from 12 to 24 hours post synchronization, times in which the cellular redox state is increased. Interestingly, the circadian clock disruption through Bmal1 knockdown on T98G cells exhibited a marked 6 hours-phase advance in the temporal response to Bortezomib compared to control cells [161]. Also, T98G cells treated with the synthetic REV-ERB agonist SR9009 showed significant differences in cell viability across time, exhibiting the lowest response to the treatment at 6 hours post synchronization. Moreover, the combined treatment of SR9009 with Bortezomib further potentiates their cytotoxic effects, clearly demonstrating a significant synergic impact of the drug combination. Since both chemotherapeutics act on different cellular targets, Bortezomib inhibits the proteasome activity while SR9009 acts on the clock-related cellular metabolism, the combined treatment should be considered as a chemotherapeutic approach for GBM cells [241].Another in vitro study reported a rhythmic pattern of p38 MAPK activity in glial cells while its levels were arrhythmic and high in IM3 glioma cells. VX-745, an inhibitor of p38 MAPK, shows an improvement to reduce glioma invasion at a specific time of day after serum shock on IM3 glioma cells [244].Recent results from our laboratory showed significant differences in Bortezomib efficacy on tumor-bearing mice when the drug was administered at the beginning of the day in the light phase or at night in the dark. We showed that a chemotherapeutic scheme in which a high dose (1.5 mg/kg) of Bortezomib was administered wholly inhibited tumor growth at both times; whereas a low dose of Bortezomib (0.5 mg/kg) displayed higher efficacy to impair tumor growth when delivered at night compared to diurnal treatment [21] (Figure 2).

Figure 2. Tumor growth inhibition (TGI) of glioma xenografts by chrono-chemotherapy using Bortezomib. C57BL/6 mice maintained in regular L/D cycles were injected with a cell line isolated from a malignant peripheral nerve sheath tumor generated in NPcis mice. Once the tumor was palpable, mice were randomly separated, and Bortezomib (0.5 mg/kg) or the vehicle, were administered at the beginning of the day or night. Tumor growth and animal weight were measured periodically. TGI of 70% and 18% was observed when Bortezomib was administered at the beginning of the night or the day, respectively [21].
References
- Sasmita, A.O.; Wong, Y.P.; Ling, A.P.K. Biomarkers and therapeutic advances in glioblastoma multiforme. Asia. Pac. J. Clin. Oncol. 2018, 14, 40–51.
- McLendon, R.; Friedman, A.; Bigner, D.; Van Meir, E.G.; Brat, D.J.; Mastrogianakis, G.M.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068.
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462.
- Huang, P.H.; Xu, A.M.; White, F.M. Oncogenic EGFR signaling networks in glioma. Sci. Signal. 2009, 2.
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772.
- Arcella, A.; Limanaqi, F.; Ferese, R.; Biagioni, F.; Oliva, M.A.; Storto, M.; Fanelli, M.; Gambardella, S.; Fornai, F. Dissecting molecular features of gliomas: Genetic loci and validated biomarkers. Int. J. Mol. Sci. 2020, 21, 685.
- De Vleeschouwer, S. (Ed.) Glioblastoma; Codon Publications: Brisbane, Australia, 2017; ISBN 9780994438126.
- Laezza, C.; D’Alessandro, A.; Di Croce, L.; Picardi, P.; Ciaglia, E.; Pisanti, S.; Malfitano, A.M.; Comegna, M.; Faraonio, R.; Gazzerro, P.; et al. P53 regulates the mevalonate pathway in human glioblastoma multiforme. Cell Death Dis. 2015, 6, e1909.
- Verreault, M.; Schmitt, C.; Goldwirt, L.; Pelton, K.; Haidar, S.; Levasseur, C.; Guehennec, J.; Knoff, D.; Labussière, M.; Marie, Y.; et al. Preclinical efficacy of the MDM2 inhibitor RG7112 in MDM2-amplified and TP53 wild-type glioblastomas. Clin. Cancer Res. 2016, 22, 1185–1196.
- Stokoe, D. PTEN. Curr. Biol. 2001, 11, R502.
- El-Khayat, S.M.; Arafat, W.O. Therapeutic strategies of recurrent glioblastoma and its molecular pathways “Lock up the beast”. Ecancermedicalscience 2021, 15, 1176.
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.L.; Burkhard, C.; Schüler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic pathways to glioblastoma: A population-based study. Cancer Res. 2004, 64, 6892–6899.
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773.
- Picca, A.; Berzero, G.; Di Stefano, A.L.; Sanson, M. The clinical use of IDH1 and IDH2 mutations in gliomas. Expert Rev. Mol. Diagn. 2018, 18, 1041–1051.
- Mondesir, J.; Willekens, C.; Touat, M.; de Botton, S. IDH1 and IDH2 mutations as novel therapeutic targets: Current perspectives. J. Blood Med. 2016, 7, 171–180.
- Jiao, Y.; Killela, P.J.; Reitman, Z.J.; Rasheed, B.A.; Heaphy, C.M.; de Wilde, R.F.; Rodriguez, F.J.; Rosemberg, S.; Oba-Shinjo, S.M.; Marie, S.K.N.; et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 2012, 3, 709–722.
- Nikiforova, M.N.; Hamilton, R.L. Molecular diagnostics of gliomas. Arch. Pathol. Lab. Med. 2011, 135, 558–568.
- Yang, P.; Zhang, W.; Wang, Y.; Peng, X.; Chen, B.; Qiu, X.; Li, G.; Li, S.; Wu, C.; Yao, K.; et al. IDH mutation and MGMT promoter methylation in glioblastoma: Results of a prospective registry. Oncotarget 2015, 6, 40896–40906.
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820.
- Oronsky, B.; Reid, T.R.; Oronsky, A.; Sandhu, N.; Knox, S.J. A Review of Newly Diagnosed Glioblastoma. Front. Oncol. 2021, 10.
- Safa, A.R.; Saadatzadeh, M.R.; Cohen-Gadol, A.A.; Pollok, K.E.; Bijangi-Vishehsaraei, K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis. 2015, 2, 152–163.
- Garnett, J.; Chumbalkar, V.; Vaillant, B.; Gururaj, A.E.; Hill, K.S.; Latha, K.; Yao, J.; Priebe, W.; Colman, H.; Elferink, L.A.; et al. Regulation of HGF expression by δEGFR-mediated c-Met activation in glioblastoma cells. Neoplasia 2013, 15, 73–84.
- Silantyev, A.S.; Falzone, L.; Libra, M.; Gurina, O.I.; Kardashova, K.S.; Nikolouzakis, T.K.; Nosyrev, A.E.; Sutton, C.W.; Mitsias, P.D.; Tsatsakis, A. Current and Future Trends on Diagnosis and Prognosis of Glioblastoma: From Molecular Biology to Proteomics. Cells 2019, 8, 863.
- Montemurro, N. Glioblastoma Multiforme and Genetic Mutations: The Issue Is Not Over Yet. An Overview of the Current Literature. J. Neurol. Surg. A 2020, 81, 64–70.
- Eoli, M.; Menghi, F.; Bruzzone, M.G.; De Simone, T.; Valletta, L.; Pollo, B.; Bissola, L.; Silvani, A.; Bianchessi, D.; D’Incerti, L.; et al. Methylation of O6-methylguanine DNA methytransferase and loss of heterozygosity on 19q and/or 17p are overlapping features of secondary glioblastomas with prolonged survival. Clin. Cancer Res. 2007, 13, 2606–2613.
- Szopa, W.; Burley, T.A.; Kramer-Marek, G.; Kaspera, W. Diagnostic and therapeutic biomarkers in glioblastoma: Current status and future perspectives. Biomed. Res. Int. 2017, 2017.
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110.
- Llaguno, S.R.A.; Parada, L.F. Cell of origin of glioma: Biological and clinical implications. Br. J. Cancer 2016, 115, 1445–1450.
- Gómez-Oliva, R.; Domínguez-García, S.; Carrascal, L.; Abalos-Martínez, J.; Pardillo-Díaz, R.; Verástegui, C.; Castro, C.; Nunez-Abades, P.; Geribaldi-Doldán, N. Evolution of Experimental Models in the Study of Glioblastoma: Toward Finding Efficient Treatments. Front. Oncol. 2021, 10.
- Jovčevska, I. Genetic secrets of long-term glioblastoma survivors. Bosn. J. Basic Med. Sci. 2019, 19, 116–124.
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563.
- Steed, T.C.; Treiber, J.M.; Patel, K.; Ramakrishnan, V.; Merk, A.; Smith, A.R.; Carter, B.S.; Dale, A.M.; Chow, L.M.L.; Chen, C.C. Differential localization of glioblastoma subtype: Implications on glioblastoma pathogenesis. Oncotarget 2016, 7, 24899–24907.
- Sidaway, P. CNS cancer: Glioblastoma subtypes revisited. Nat. Rev. Clin. Oncol. 2017, 14, 587.
- Dunlap, J.C.; Loros, J.J.; DeCoursey, P.J. (Eds.) Chronobiology: Biological Timekeeping; Sinauer Associates: Sunderland, MA, USA, 2004; ISBN 087893149X.
- Takahashi, J.S.; Hong, H.K.; Ko, C.H.; McDearmon, E.L. The genetics of mammalian circadian order and disorder: Implications for physiology and disease. Nat. Rev. Genet. 2008, 9, 764–775.
- Golombek, D.A.; Rosenstein, R.E. Physiology of circadian entrainment. Physiol. Rev. 2010, 90, 1063–1102.
- Welsh, D.K.; Takahashi, J.S.; Kay, S.A. Suprachiasmatic nucleus: Cell autonomy and network properties. Annu. Rev. Physiol. 2009, 72, 551–577.
- Guido, M.E.; Goguen, D.; De Guido, L.; Robertson, H.A.; Rusak, B. Circadian and photic regulation of immediate-early gene expression in the hamster suprachiasmatic nucleus. Neuroscience 1999, 90, 555–571.
- Guido, M.E.; Garbarino-Pico, E.; Contin, M.A.; Valdez, D.J.; Nieto, P.S.; Verra, D.M.; Acosta-Rodriguez, V.A.; de Zavalía, N.; Rosenstein, R.E. Inner retinal circadian clocks and non-visual photoreceptors: Novel players in the circadian system. Prog. Neurobiol. 2010, 92, 484–504.
- Yamazaki, S.; Numano, R.; Abe, M.; Hida, A.; Takahashi, R.I.; Ueda, M.; Block, G.D.; Sakaki, Y.; Menaker, M.; Tei, H. Resetting central and peripheral circadian oscillators in transgenic rats. Science 2000, 288, 682–685.
- Huang, X.-L.; Fu, C.-J.; Bu, R.-F. Role of Circadian Clocks in the Development and Therapeutics of Cancer. J. Int. Med. Res. 2011, 39.
- Kubo, T.; Ozasa, K.; Mikami, K.; Wakai, K.; Fujino, Y.; Watanabe, Y.; Miki, T.; Nakao, M.; Hayashi, K.; Suzuki, K.; et al. Prospective cohort study of the risk of prostate cancer among rotating-shift workers: Findings from the Japan Collaborative Cohort Study. Am. J. Epidemiol. 2006, 164, 549–555.
- Masri, S.; Sassone-Corsi, P. The emerging link between cancer, metabolism, and circadian rhythms. Nat. Med. 2018, 24, 1795–1803.
- Green, C.B.; Takahashi, J.S.; Bass, J. The Meter of Metabolism. Cell 2008, 134, 728–742.
- Hardin, P.E.; Hall, J.C.; Rosbash, M. Feedback of the Drosophila period gene product on circadian cycling of its messenger RNA levels. Nature 1990, 343, 536–540.
- Buhr, E.D.; Takahashi, J.S. Molecular components of the mammalian circadian clock. Handb. Exp. Pharmacol. 2013, 217, 3–27.
- Lee, C.; Etchegaray, J.P.; Cagampang, F.R.A.; Loudon, A.S.I.; Reppert, S.M. Posttranslational mechanisms regulate the mammalian circadian clock. Cell 2001, 107, 855–867.
- Eide, E.J.; Kang, H.; Crapo, S.; Gallego, M.; Virshup, D.M. Casein kinase I in the mammalian circadian clock. Methods Enzymol. 2005, 393, 408–418.
- Godinho, S.I.H.; Maywood, E.S.; Shaw, L.; Tucci, V.; Barnard, A.R.; Busino, L.; Pagano, M.; Kendall, R.; Quwailid, M.M.; Romero, M.R.; et al. The after-hours mutant reveals a role for Fbxl3 in determining mammalian circadian period. Science 2007, 316, 897–900.
- Shirogane, T.; Jin, J.; Ang, X.L.; Harper, J.W. SCFβ-TRCP controls Clock-dependent transcription via casein kinase 1-dependent degradation of the mammalian period-1 (Per1) protein. J. Biol. Chem. 2005, 280, 26863–26872.
- Siepka, S.M.; Yoo, S.H.; Park, J.; Song, W.; Kumar, V.; Hu, Y.; Lee, C.; Takahashi, J.S. Circadian Mutant Overtime Reveals F-box Protein FBXL3 Regulation of Cryptochrome and Period Gene Expression. Cell 2007, 129, 1011–1023.
- Yoo, S.H.; Mohawk, J.A.; Siepka, S.M.; Shan, Y.; Huh, S.K.; Hong, H.K.; Kornblum, I.; Kumar, V.; Koike, N.; Xu, M.; et al. Competing E3 ubiquitin ligases govern circadian periodicity by degradation of CRY in nucleus and cytoplasm. Cell 2013, 152, 1091–1105.
- Preitner, N.; Damiola, F.; Zakany, J.; Duboule, D.; Albrecht, U.; Schibler, U. The orphan nuclear receptor REV-ERBα controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 2002, 110, 251–260.
- Sato, T.K.; Panda, S.; Miraglia, L.J.; Reyes, T.M.; Rudic, R.D.; McNamara, P.; Naik, K.A.; Fitzgerald, G.A.; Kay, S.A.; Hogenesch, J.B. A functional genomics strategy reveals rora as a component of the mammalian circadian clock. Neuron 2004, 43, 527–537.
- Sancar, G.; Brunner, M. Circadian clocks and energy metabolism. Cell. Mol. Life Sci. 2014, 71, 2667–2680.
- Fu, L.; Kettner, N.M. The circadian clock in cancer development and therapy. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2013; Volume 119, pp. 221–282. ISBN 9780123969712.
- Panda, S.; Antoch, M.P.; Miller, B.H.; Su, A.I.; Schook, A.B.; Straume, M.; Schultz, P.G.; Kay, S.A.; Takahashi, J.S.; Hogenesch, J.B. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 2002, 109, 307–320.
- Miller, B.H.; McDearmon, E.L.; Panda, S.; Hayes, K.R.; Zhang, J.; Andrews, J.L.; Antoch, M.P.; Walker, J.R.; Esser, K.A.; Hogenesch, J.B.; et al. Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc. Natl. Acad. Sci. USA 2007, 104, 3342–3347.
- Arafa, K.; Emara, M. Insights About Circadian Clock and Molecular Pathogenesis in Gliomas. Front. Oncol. 2020, 10, 199.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Sulli, G.; Lam, M.T.Y.; Panda, S. Interplay between Circadian Clock and Cancer: New Frontiers for Cancer Treatment. Trends Cancer 2019, 5, 475–494.
- Shafi, A.A.; Knudsen, K.E. Cancer and the circadian clock. Cancer Res. 2019, 79, 3806–3814.
- Soták, M.; Sumová, A.; Pácha, J. Cross-talk between the circadian clock and the cell cycle in cancer. Ann. Med. 2014, 46, 221–232.
- Shostak, A. Circadian clock, cell division, and cancer: From molecules to organism. Int. J. Mol. Sci. 2017, 18, 873.
- Farshadi, E.; Yan, J.; Leclere, P.; Goldbeter, A.; Chaves, I.; van der Horst, G.T.J. The positive circadian regulators CLOCK and BMAL1 control G2/M cell cycle transition through Cyclin B1. Cell Cycle 2019, 18, 16–33.
- Kiessling, S.; Beaulieu-Laroche, L.; Blum, I.D.; Landgraf, D.; Welsh, D.K.; Storch, K.F.; Labrecque, N.; Cermakian, N. Enhancing circadian clock function in cancer cells inhibits tumor growth. BMC Biol. 2017, 15, 13.
- Hoffman, A.E.; Zheng, T.; Ba, Y.; Stevens, R.G.; Yi, C.H.; Leaderer, D.; Zhu, Y. Phenotypic effects of the circadian gene Cryptochrome 2 on cancer-related pathways. BMC Cancer 2010, 10, 110.
- Lee, S.; Donehower, L.A.; Herron, A.J.; Moore, D.D.; Fu, L. Disrupting circadian homeostasis of sympathetic signaling promotes tumor development in mice. PLoS ONE 2010, 5, e10995.
- Fu, L.; Pelicano, H.; Liu, J.; Huang, P.; Lee, C.C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 2002, 111, 41–50.
- Gotoh, T.; Vila-Caballer, M.; Santos, C.S.; Liu, J.; Yang, J.; Finkielstein, C.V. The circadian factor Period 2 modulates p53 stability and transcriptional activity in unstressed cells. Mol. Biol. Cell 2014, 25, 3081–3093.
- Gotoh, T.; Kim, J.K.; Liu, J.; Vila-Caballer, M.; Stauffer, P.E.; Tyson, J.J.; Finkielstein, C.V. Model-driven experimental approach reveals the complex regulatory distribution of p53 by the circadian factor period 2. Proc. Natl. Acad. Sci. USA 2016, 113, 13516–13521.
- Gotoh, T.; Vila-Caballer, M.; Liu, J.; Schiffhauer, S.; Finkielstein, C.V. Association of the circadian factor Period 2 to p53 influences p53’s function in DNA-damage signaling. Mol. Biol. Cell 2015, 26, 359–372.
- Zhanfeng, N.; Chengquan, W.; Hechun, X.; Jun, W.; Lijian, Z.; Dede, M.; Wenbin, L.; Lei, Y. Period2 downregulation inhibits glioma cell apoptosis by activating the MDM2-TP53 pathway. Oncotarget 2016, 7, 27350–27362.
- Relógio, A.; Thomas, P.; Medina-Pérez, P.; Reischl, S.; Bervoets, S.; Gloc, E.; Riemer, P.; Mang-Fatehi, S.; Maier, B.; Schäfer, R.; et al. Ras-Mediated Deregulation of the Circadian Clock in Cancer. PLoS Genet. 2014, 10, e1004338.
- El-Athman, R.; Genov, N.N.; Mazuch, J.; Zhang, K.; Yu, Y.; Fuhr, L.; Abreu, M.; Li, Y.; Wallach, T.; Kramer, A.; et al. The Ink4a/Arf locus operates as a regulator of the circadian clock modulating RAS activity. PLoS Biol. 2017, 15, e2002940.
- Wagner, P.M.; Sosa Alderete, L.G.; Gorné, L.D.; Gaveglio, V.; Salvador, G.; Pasquaré, S.; Guido, M.E. Proliferative Glioblastoma Cancer Cells Exhibit Persisting Temporal Control of Metabolism and Display Differential Temporal Drug Susceptibility in Chemotherapy. Mol. Neurobiol. 2019, 56, 1276–1292.
- Marquez, S.; Crespo, P.; Carlini, V.; Garbarino-Pico, E.; Baler, R.; Caputto, B.L.; Guido, M.E. The metabolism of phospholipids oscillates rhythmically in cultures of fibroblasts and is regulated by the clock protein PERIOD 1. FASEB J. 2004, 18, 519–521.
- Verlande, A.; Masri, S. Circadian Clocks and Cancer: Timekeeping Governs Cellular Metabolism. Trends Endocrinol. Metab. 2019, 30, 445–458.
- Méndez, I.; Díaz-Muñoz, M. Circadian and metabolic perspectives in the role played by NADPH in cancer. Front. Endocrinol. 2018, 9, 15.
- Dyar, K.A.; Lutter, D.; Artati, A.; Ceglia, N.J.; Liu, Y.; Armenta, D.; Jastroch, M.; Schneider, S.; de Mateo, S.; Cervantes, M.; et al. Atlas of Circadian Metabolism Reveals System-wide Coordination and Communication between Clocks. Cell 2018, 174, 1571.e11–1585.e11.
- Vollmers, C.; Gill, S.; DiTacchio, L.; Pulivarthy, S.R.; Le, H.D.; Panda, S. Time of feeding and the intrinsic circadian clock drive rhythms in hepatic gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 21453–21458.
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47.
- Scheiermann, C.; Gibbs, J.; Ince, L.; Loudon, A. Clocking in to immunity. Nat. Rev. Immunol. 2018, 18, 423–437.
- Hadadi, E.; Acloque, H. Role of circadian rhythm disorders on EMT and tumour-immune interactions in endocrine-related cancers. Endocr. Relat. Cancer 2021, 28, R67–R80.
- Matsunaga, N.; Kohno, Y.; Kakimoto, K.; Hayashi, A.; Koyanagi, S.; Ohdo, S. Influence of CLOCK on cytotoxicity induced by diethylnitrosamine in mouse primary hepatocytes. Toxicology 2011, 280, 144–151.
- Wu, Y.; Sato, F.; Bhawal, U.K.; Kawamoto, T.; Fujimoto, K.; Noshiro, M.; Seino, H.; Morohashi, S.; Kato, Y.; Kijima, H. BHLH transcription factor DEC2 regulates pro-apoptotic factor bim in human oral cancer HSC-3 cells. Biomed. Res. 2012, 33, 75–82.
- Wang, J.; Mauvoisin, D.; Martin, E.; Atger, F.; Galindo, A.N.; Dayon, L.; Sizzano, F.; Palini, A.; Kussmann, M.; Waridel, P.; et al. Nuclear Proteomics Uncovers Diurnal Regulatory Landscapes in Mouse Liver. Cell Metab. 2017, 25, 102–117.
- Gery, S.; Komatsu, N.; Baldjyan, L.; Yu, A.; Koo, D.; Koeffler, H.P. The Circadian Gene Per1 Plays an Important Role in Cell Growth and DNA Damage Control in Human Cancer Cells. Mol. Cell 2006, 22, 375–382.
- Kang, T.H.; Leem, S.H. Modulation of ATR-mediated DNA damage checkpoint response by cryptochrome 1. Nucleic Acids Res. 2014, 42, 4427–4434.
- Kang, T.H.; Reardon, J.T.; Kemp, M.; Sancar, A. Orcadian oscillation of nucleotide excision repair in mammalian brain. Proc. Natl. Acad. Sci. USA 2009, 106, 2864–2867.
- Papp, S.J.; Huber, A.L.; Jordan, S.D.; Kriebs, A.; Nguyen, M.; Moresco, J.J.; Yates, J.R.; Lamia, K.A. DNA damage shifts circadian clock time via Hausp-dependent Cry1 stabilization. eLife 2015, 4, e04883.
- Oklejewicz, M.; Destici, E.; Tamanini, F.; Hut, R.A.; Janssens, R.; van der Horst, G.T.J. Phase Resetting of the Mammalian Circadian Clock by DNA Damage. Curr. Biol. 2008, 18, 286–291.
- Kolinjivadi, A.M.; Chong, S.T.; Ngeow, J. Molecular connections between circadian rhythm and genome maintenance pathways. Endocr. Relat. Cancer 2021, 28, R55–R66.
- Dong, Z.; Zhang, G.; Qu, M.; Gimple, R.C.; Wu, Q.; Qiu, Z.; Prager, B.C.; Wang, X.; Kim, L.J.Y.; Morton, A.R.; et al. Targeting glioblastoma stem cells through disruption of the circadian clock. Cancer Discov. 2019, 9, 1556–1573.
- Li, A.; Lin, X.; Tan, X.; Yin, B.; Han, W.; Zhao, J.; Yuan, J.; Qiang, B.; Peng, X. Circadian gene clock contributes to cell proliferation and migration of glioma and is directly regulated by tumor-suppressive miR-124. FEBS Lett. 2013, 587, 2455–2460.
- Chang, W.H.; Lai, A.G. Timing gone awry: Distinct tumour suppressive and oncogenic roles of the circadian clock and crosstalk with hypoxia signalling in diverse malignancies. J. Transl. Med. 2019, 17, 132.
- Chen, P.; Hsu, W.H.; Chang, A.; Tan, Z.; Lan, Z.; Zhou, A.; Spring, D.J.; Lang, F.F.; Wang, Y.A.; Depinho, R.A. Circadian regulator CLOCK recruits immune-suppressive microglia into the GBM tumor microenvironment. Cancer Discov. 2020, 10, 371–381.
- Crespo, I.; Tão, H.; Nieto, A.B.; Rebelo, O.; Domingues, P.; Vital, A.L.; Patino, M.D.C.; Barbosa, M.; Lopes, M.C.; Oliveira, C.R.; et al. Amplified and Homozygously Deleted Genes in Glioblastoma: Impact on Gene Expression Levels. PLoS ONE 2012, 7, e46088.
- Chen, Z.; Liu, P.; Li, C.; Luo, Y.; Chen, I.; Liang, W.; Chen, X.; Feng, Y.; Xia, H.; Wang, F. Deregulated expression of the clock genes in gliomas. Technol. Cancer Res. Treat. 2013, 12, 91–97.
- Madden, M.H.; Anic, G.M.; Thompson, R.C.; Nabors, L.B.; Olson, J.J.; Browning, J.E.; Monteiro, A.N.; Egan, K.M. Circadian pathway genes in relation to glioma risk and outcome. Cancer Causes Control 2014, 25, 25–32.
- Wang, F.; Li, C.; Chen, L. The Circadian Gene Clock Plays an Important Role in Cell Apoptosis and the DNA Damage Response In Vitro. Technol. Cancer Res. Treat. 2016, 15, 480–486.
- Wang, Z.; Su, G.; Dai, Z.; Meng, M.; Zhang, H.; Fan, F.; Liu, Z.; Zhang, L.; Weygant, N.; He, F.; et al. Circadian clock genes promote glioma progression by affecting tumour immune infiltration and tumour cell proliferation. Cell Prolif. 2021, 54, e12988.
- Wagner, P.M.; Prucca, C.G.; Velazquez, F.N.; Sosa Alderete, L.G.; Caputto, B.L.; Guido, M.E. Temporal regulation of tumor growth in nocturnal mammals: In vivo studies and chemotherapeutical potential. FASEB J. 2021, 35, e21231.
- Slat, E.A.; Sponagel, J.; Marpegan, L.; Simon, T.; Kfoury, N.; Kim, A.; Binz, A.; Herzog, E.D.; Rubin, J.B. Cell-intrinsic, Bmal1-dependent Circadian Regulation of Temozolomide Sensitivity in Glioblastoma. J. Biol. Rhythm. 2017, 32, 121–129.
- Jung, C.H.; Kim, E.M.; Park, J.K.; Hwang, S.G.; Moon, S.K.; Kim, W.J.; Um, H.D. Bmal1 suppresses cancer cell invasion by blocking the phosphoinositide 3-kinase-Akt-MMP-2 signaling pathway. Oncol. Rep. 2013, 29, 2109–2113.
- Gwon, D.H.; Lee, W.Y.; Shin, N.; Kim, S.I.; Jeong, K.; Lee, W.H.; Kim, D.W.; Hong, J.; Lee, S.Y. BMAL1 suppresses proliferation, migration, and invasion of U87MG cells by downregulating cyclin b1, phospho-AKT, and metalloproteinase-9. Int. J. Mol. Sci. 2020, 21, 2352.
- Khan, S.; Liu, Y.; Siddique, R.; Nabi, G.; Xue, M.; Hou, H. Impact of chronically alternating light-dark cycles on circadian clock mediated expression of cancer (Glioma)-related genes in the brain. Int. J. Biol. Sci. 2019, 15, 1816–1834.
- Xia, H.C.; Niu, Z.F.; Ma, H.; Cao, S.Z.; Hao, S.C.; Liu, Z.T.; Wang, F. Deregulated expression of the Per1 and Per2 in human gliomas. Can. J. Neurol. Sci. 2010, 37, 365–370.
- Zhu, L.; Wang, Q.; Hu, Y.; Wang, F. The circadian gene PER1 plays an important role in radiation-induced apoptosis and DNA damage in glioma. Asian Pac. J. Cancer Prev. 2019, 20, 2195–2201.
- Zhanfeng, N.; Yanhui, L.; Zhou, F.; Shaocai, H.; Guangxing, L.; Hechun, X. Circadian genes Per1 and Per2 increase radiosensitivity of glioma in vivo. Oncotarget 2015, 6, 9951–9958.
- Gao, Y.; Wu, Y.; Zhang, N.; Yuan, H.; Wang, F.; Xu, H.; Yu, J.; Ma, J.; Hou, S.; Cao, X. IDH1 gene mutation activates Smad signaling molecules to regulate the expression levels of cell cycle and biological rhythm genes in human glioma U87-MG cells. Mol. Med. Rep. 2021, 23.
- Wang, F.; Luo, Y.; Li, C.; Chen, L. Correlation between deregulated expression of PER2 gene and degree of glioma malignancy. Tumori 2014, 100, e266–e272.
- Ma, D.; Hou, L.; Xia, H.; Li, H.; Fan, H.; Jia, X.; Niu, Z. PER2 inhibits proliferation and stemness of glioma stem cells via the Wnt/ß-catenin signaling pathway. Oncol. Rep. 2020, 44, 533–542.
- Luo, Y.; Wang, F.; Chen, L.A.; Chen, X.W.; Chen, Z.J.; Liu, P.F.; Li, F.F.; Li, C.Y.; Liang, W. Deregulated expression of Cry1 and Cry2 in human gliomas. Asian Pac. J. Cancer Prev. 2012, 13, 5725–5728.
- Fan, W.; Caiyan, L.; Ling, Z.; Jiayun, Z. Aberrant rhythmic expression of cryptochrome2 regulates the radiosensitivity of rat gliomas. Oncotarget 2017, 8, 77809–77818.
- Yu, M.; Li, W.; Wang, Q.; Wang, Y.; Lu, F. Circadian regulator NR1D2 regulates glioblastoma cell proliferation and motility. Oncogene 2018, 37, 4838–4853.
- Wagner, P.M.; Monjes, N.M.; Guido, M.E. Chemotherapeutic Effect of SR9009, a REV-ERB Agonist, on the Human Glioblastoma T98G Cells. ASN Neuro 2019, 11.
- Sulli, G.; Rommel, A.; Wang, X.; Kolar, M.J.; Puca, F.; Saghatelian, A.; Plikus, M.V.; Verma, I.M.; Panda, S. Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature 2018, 553, 351–355.
- Wang, F.; Chen, Q.X. The analysis of deregulated expression of the timeless genes in gliomas. J. Cancer Res. Ther. 2018, 14, S708–S712.
- Razorenova, O.V. Brain and muscle ARNT-like protein BMAL1 regulates ROS homeostasis and senescence: A possible link to hypoxia-inducible factor-mediated pathway. Cell Cycle 2012, 11, 213.
- Hatanaka, F.; Matsubara, C.; Myung, J.; Yoritaka, T.; Kamimura, N.; Tsutsumi, S.; Kanai, A.; Suzuki, Y.; Sassone-Corsi, P.; Aburatani, H.; et al. Genome-Wide Profiling of the Core Clock Protein BMAL1 Targets Reveals a Strict Relationship with Metabolism. Mol. Cell. Biol. 2010, 30, 5636–5648.
- Khapre, R.V.; Kondratova, A.A.; Susova, O.; Kondratov, R.V. Circadian clock protein BMAL1 regulates cellular senescence in vivo. Cell Cycle 2011, 10, 4162–4169.
- Cho, H.; Zhao, X.; Hatori, M.; Yu, R.T.; Barish, G.D.; Lam, M.T.; Chong, L.W.; Ditacchio, L.; Atkins, A.R.; Glass, C.K.; et al. Regulation of circadian behaviour and metabolism by REV-ERB-α and REV-ERB-β. Nature 2012, 485, 123–127.
- Solt, L.A.; Wang, Y.; Banerjee, S.; Hughes, T.; Kojetin, D.J.; Lundasen, T.; Shin, Y.; Liu, J.; Cameron, M.D.; Noel, R.; et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature 2012, 485, 62–68.
- Woldt, E.; Sebti, Y.; Solt, L.A.; Duhem, C.; Lancel, S.; Eeckhoute, J.; Hesselink, M.K.C.; Paquet, C.; Delhaye, S.; Shin, Y.; et al. Rev-erb-α modulates skeletal muscle oxidative capacity by regulating mitochondrial biogenesis and autophagy. Nat. Med. 2013, 19, 1039–1046.
- Vieira, E.; Marroquí, L.; Batista, T.M.; Caballero-Garrido, E.; Carneiro, E.M.; Boschero, A.C.; Nadal, A.; Quesada, I. The clock gene Rev-erbα regulates pancreatic β-cell function: Modulation by leptin and high-fat diet. Endocrinology 2012, 153, 592–601.
- Scheiermann, C.; Kunisaki, Y.; Frenette, P.S. Circadian control of the immune system. Nat. Rev. Immunol. 2013, 13, 190–198.


