1. GBM General Hallmarks
Gliomagenesis, as a multi-component process that promotes the development of gliomas, involves amplification and deletion or mutation of several genes, including the epidermal growth factor receptor (EGFR), tumor protein 53 (TP53), phosphate and tensin homolog (PTEN), and isocitrate dehydrogenase (IDH), among others [
23]. These genes regulate distinct pathways known to be part of the core drivers of gliomagenesis, leading to aberrant signaling in proliferation, cell cycle regulation, senescence, and apoptosis [
24,
25].
One of the most studied hallmarks of human GBM is the amplification and genetic rearrangement of the gene that encodes for the tyrosine kinase receptor known as EGFR. This pathway can be activated either through overexpression of the receptor, amplification of the EGFR locus, and/or mutations in the receptor [
26]. The most common and described mutation in GBM is the EGFRvIII, which corresponds to the loss of exons 2–7, resulting in a truncated extracellular domain with ligand-independent constitutive activity and consequently excessive cell proliferation. This mutation is associated with a bad prognosis and has not been observed in healthy tissues and secondary GBM subtypes [
27]. Interestingly, GBM cells express either EGFR or EGFRvIII, although co-expression of both variants has also been reported in a small population of cells [
2]. The TP53 gene encodes for a tumor suppressor protein that participates in cell cycle control, DNA damage response, cell death, and differentiation. Its mutation incidence is low in primary tumors (about 30%); however, 90% of secondary GBMs present mutations in this gene. Indeed, it has been proposed that TP53 mutation is an early event in secondary GBM [
6] and is correlated with GBM progression by driving the activation of the mevalonate pathway since p53-mutant cells have shown an elevated activity of this pathway compared to wild type cells [
28]. Other alterations related to this pathway include murine double minute 2 (MDM2) and MDM4 amplification, and CDKN2A-p14ARF deletion [
24]. As MDM2 is a negative regulator of the TP53 gene, using inhibitors of MDM2 has shown promising results in GBM treatment [
29]. PTEN gene is another commonly mutated tumor suppressor gene observed in most cancers, similar to TP53 [
30]. It has a crucial role in inhibiting cell proliferation and regulating the migration and invasion of cells. PTEN is frequently inactivated in GBM, either by losing heterozygosity (LOH) of chromosome 10 or by mutation-induced constitutive activation of PI3K. The LOH of chromosome 10 is observed in almost 70% of GBM samples, predominantly in the primary subtype [
23]. Amplification of platelet-derived growth factor receptor (PDGFR) is another genetic alteration observed in GBM tumors [
31]. Lastly, IDH mutations are considered the most reliable indicator to differentiate primary from secondary GBM [
32,
33,
34,
35,
36], primary GBM typically lacking IDH mutations [
37]. The IDH gene encodes for isocitrate dehydrogenase, an enzyme that catalyzes the oxidative decarboxylation of isocitrate to 2-oxoglutarate within the Krebs cycle, whereas IDH mutants catalyze the production of the oncometabolite 2-hydroxyglutarate (2-HG). Importantly, patients treated with TMZ are associated with a favorable prognosis when they present IDH mutations since the synthesis of 2-HG interferes in the activation of DNA demethylation enzymes, yielding a hypermethylation status in tumor cells [
38].
The O6-methylguanine-DNA methyltransferase (O6-MGMT) gene encodes for an enzyme that removes the methyl group from the guanine (position O6). The expression level of this protein is relevant to the treatment outcome when using TMZ since its expression is associated with a poor response. Consequently, the methylation level of its promoter is associated with a better response to TMZ treatment [
23].
2. GBM Classification
The WHO classifies brain tumors into four groups (I–IV) of growing malignancy based on the morphological features of the tumor and their cells of origin [
39]. Grades I and II include tumors with low proliferation potential, whereas grades III and IV tumors are high-grade gliomas characterized by high proliferation rates and aggressiveness [
40]. GBM is classified as a grade IV high-speed growth tumor showing diffuse boundaries and is usually associated with a poor prognosis [
6]. GBM is also divided into primary and secondary tumors, with primary GBM commonly diagnosed in the elderly without prior disease. Several alterations have been reported in primary GBM including LOH at 10q (70% of cases) [
27,
32,
41] and 10p (50–70%) [
27,
42], amplification or mutation of EGFR (~35–45%) [
27,
32,
41,
42,
43] mutation in TP53 (27–30%) [
27,
43], deletion of cyclin-dependent kinase inhibitor 2A/B (CDKN2A/B) (31%) [
44], mutation or deletion of PTEN (25%) [
27], promoter methylation of O6-MGMT (42%) [
45], promoter mutation of telomerase reverse transcriptase (TERT) (72%) [
41,
43], mutation in glioma-associated oncogene homolog 1 (GLI1) (5–22%), deletion or mutation in phosphatidylinositol-4,5-bisphosphate 3-kinase A (PIK3CA) (~1%), MDM2 (7–12%) [
41,
42,
44] and neurofibromatosis type1 (NF1) (11%), amplification of PDGFR (7%), and mutation in IDH1/2 (5%) [
44].
On the other hand, secondary GBM develops from a low-grade glioma or an anaplastic astrocytoma, affects younger persons, and shows a better prognosis after diagnosis. These tumors are much less common, showing genetic alterations that include mutation in IDH1/2 (73–85%) [
46], TP53 (65–81%), ATRX (~65–71%) [
27,
43], and PTEN (<5%) [
27] genes, promoter methylation of MGMT (79%) [
45], loss of chromosome 19q (~50%) and 10q (63%) [
27], p16
INK4a deletion (19%) and EGFR amplification (8%) [
32]. Mutant IDH1 is considered a metabolic marker of secondary GBM because of its ubiquitous expression in lower-grade gliomas that eventually progress to GBM. Besides the differences described above regarding genetic profiles, primary and secondary GBMs are histologically indistinguishable, and the most reliable indicator to differentiate them are mutations in the IDH1 gene [
36]. A summary of the most frequent genetic alterations in primary and secondary GBMs is presented in
Table 1.
Table 1. Genetic alterations of primary and secondary Glioblastoma (GBM).
Primary GBM
[27,43,44,45,46] |
Secondary GBM
[27,32,45,46] |
| LOH chromosome 10q (70%) |
IDH1/2 mutation (73–85%) |
| LOH chromosome 10p (50–70%) |
TP53 mutation (65–81%) |
| EGFR amplification or mutation (35–45%) |
ATRX mutation (65–71%) |
| TP53 mutation (27–30%) |
LOH chromosome 10q (63%) |
| PTEN mutation (25%) |
LOH chromosome 19q (~50%) |
| O6-MGMT promoter methylation (42%) |
MGMT promoter methylation (79%) |
| TERT promoter mutation (72%) |
p16INK4a deletion (~19%) |
| PDGFR amplification (~7%) |
EGFR amplification (8%) |
| MDM2 mutation (7–12%) |
PTEN mutation (<5%) |
| NF1 mutation/deletion (11%) |
|
| GLI1 mutation (5–22%) |
|
| IDH1/2 mutation (5%) |
|
| PIK3CA mutation (1%) |
|
In 2016, GBM classification was updated, considering the specific molecular and genetic profiles observed in the different tumors [
25,
47]. Based on this, GBM was classified into four subtypes: proneural, neural, classic, and mesenchymal [
6,
23,
48,
49,
50]. The proneural group constitutes the most frequent secondary GBM and has histological features most consistent with oligodendrocytes. This subtype, typically found in younger patients, is associated with the best prognosis after treatment. The most frequent genetic alterations observed in this subtype of GBM are mutations in PDGFRA, IDH1, TP53, and PIK3C genes. The neural profile is characterized by TP53 mutation, EGFR amplification, and CDKN2A deletion. The histology that describes this subtype is consistent with a combination of oligodendroglial, astrocytic, and neuronal features. Also, an important composition of genes involved in nervous system development and function (NEFL, GABRA1, SYT1, and SLC12A5) and a greater degree of neuronal marker expression was observed in the neural subtype. The classic or proliferative subtype is associated with EGFR amplification (97%), LOH of chromosome 10, chromosome 7 amplification, and CDKN2A-p16
INK4a deletion (94%) and demonstrated features more consistent with astrocytes. The mesenchymal subtype of GBM is associated with a worse prognosis and evidence of a greater degree of necrosis and inflammatory components. This profile is characterized by overexpression of mesenchymal and astrocytic markers, lower expression of the tumor suppressor NF1, altered PTEN, TP53, CDKN2A, Akt genes, and the presence of mesenchymal markers (MET, CHI3L1, CD44, and MERTK). The classical and mesenchymal subtypes are associated with more aggressive high-grade gliomas, the worst prognosis compared to other profiles, and a slightly better response to aggressive therapies [
23,
47,
48,
50,
51].
Table 2 summarizes the most important features of the four subtypes described above.
Table 2. Features of neural, proneural, classical, and mesenchymal GBM subtypes.
| GBM Subtype |
Molecular and Genetic Profile |
Median Survival
(Months) |
Proneural
[6,23,47,48,50] |
- -
-
IDH1 point mutation
- -
-
PDGFRA alterations
- -
-
TP53, DLL3, DCX, TCF4, SOX, ASCL1, OLIG2 mutations
- -
-
PIK3C mutation
- -
-
Expression of NKX2-2
- -
-
Associated to secondary GBM
|
11.3 (9.3–14.7) |
Neural
[23,47,50] |
- -
-
Expression of neuron markers (NEFL, GABRA, SYT1, and SLC12A5)
|
13.1 (9.8–18) |
Classical
[23,47,48,50] |
- -
-
EGFR amplification
- -
-
Chromosome 7 amplification
- -
-
LOH 10
- -
-
CDKN2A deletion
- -
-
High Notch and Sonic Hedgehog genes expression
- -
-
NES expression
|
12.2 (11.08–18) |
Mesenchymal
[23,47,48,50] |
- -
-
Lower expression of NF1 PTEN, TP53, CDKN2A, Akt alterations
- -
-
Expression of mesenchymal markers (MET, CHI3L1, CD44, and MERTK)
- -
-
Expression of SERPINE, TRADD, RELB, CTGF and TNFRS1A.
- -
-
Focal deletions 17q11.2
|
11.8 (9.57–15.4) |
In addition to the molecular and genetic features, the different subtypes of GBM have also been associated with the distinct localization of the tumors in the brain. Regarding the anatomical localization of the different subtypes, it was reported that tumors belonging to proneural and neural subtypes are found in the subventricular zone (SVZ) and showed a more rapid progression and poorer response to treatment compared to those localized outside of the SVZ of the brain. On the contrary, classical and mesenchymal GBMs are localized diffusely and away from the SVZ [
52]. Interestingly, a recent transcriptome analysis revealed only three subtypes of GBM, presenting strongly enriched mRNAs associated with classical, proneural, and mesenchymal subtypes. This observation suggests that the neural subtype may represent a contamination of the original samples with non-tumor cells [
53]. Regarding the response of the different GBM subtypes to treatment, it was observed that an aggressive therapeutic approach significantly reduces the mortality of patients with tumors belonging to the classical and mesenchymal subtypes. On the contrary, patients diagnosed with the neural subtype showed a slight response to this treatment, and non-significant changes were observed in the proneural patient cohort [
47].
3. Circadian Rhythms
The circadian clocks (from Latin
circa: near/
dies: day) temporarily regulate cell-autonomous oscillations with a 24-h periodicity of a large array of biological processes and behaviors such as sleep/wake cycles, feeding/fasting control, metabolism, hormone secretion, and immune function [
20]. The evolutionarily conserved circadian mechanism in the diverse species studied (reviewed in [
120]) is made up of central and peripheral oscillators distributed in organs, tissues, and even in individual cells [
20]. The suprachiasmatic nuclei (SCN) in the anterior hypothalamus harbors the master circadian clock, which is synchronized by external cues, or
Zeitgebers (timer-givers) such as light or temperature, among others to anticipate and adapt the circadian timekeeping system to the environment [
121,
122,
123].
Light and the environmental illumination conditions are the strongest synchronizers of the SCN through the projections from the retina [
121,
124]. The master clock can coordinate circadian outputs to synchronize peripheral clocks (e.g., liver, kidney, skin, intestine, lung, pancreas, ovary, and heart) in a tissue-specific manner through the autonomic nervous and the neuroendocrine systems [
122,
125]. Therefore, the central clock and peripheral oscillators drive the rhythmic expression of genes to couple physiological and behavioral processes to periodic environmental changes. However, modern life characterized by increased night-time activities with prolonged artificial lighting such as rotating shift work, hypercaloric diets, shortened sleep hours, and jet lag alters endogenous homeostasis with external cues. Consequently, this misalignment characterized by loss of the correct coordination between elements of the circadian system is considered a contributing factor to the development of metabolic syndrome, inflammatory disorders, and higher cancer risk [
126,
127,
128,
129].
4. The Molecular Clock
In 2017, the Nobel Prize in Physiology and Medicine was awarded to J.C. Hall, M. Rosbash, and M.W. Young for describing the molecular clock mechanism that underlies the circadian rhythms using fruit flies as a model organism. They showed that a gene named Period encodes for a protein whose expression was regulated by a negative feedback loop [
130]. Later, other proteins of the circadian machinery were identified and extended to other species to elucidate the molecular clockwork mechanism in the cell. In mammals, the molecular clock comprises the so-called transcriptional/translational feedback loops (TTFL) [
120,
131] including a core set of
clock genes that encodes positive and negative regulators. The primary loop involves the positive elements
Bmal1 (aryl hydrocarbon receptor nuclear translocator-like,
Arntl) and
Clock (circadian locomotor output cycles kaput) and its paralogue neuronal PAS domain protein 2,
Npas2, and the negative components
Per1/2 (Period) and
Cry1/2 (Cryptochrome) genes. During the day, the CLOCK:BMAL1 heterodimer recognizes the E-box sequence in
Per and
Cry promoters, increasing the levels of these transcripts. Then, PER and CRY proteins form a repressor complex that translocates to the nucleus and represses the CLOCK:BMAL1 activity inhibiting their expression. During the night, the repressor complex of PER and CRY is degraded, allowing CLOCK:BMAL1 to activate a new cycle of transcription and translation of approximately 24 h [
132].
Furthermore, post-translational modifications of PER and CRY proteins such as phosphorylation-dependent ubiquitination and proteasomal degradation occur in a circadian manner and regulate the subcellular localization and/or half-life of proteins contributing to the progression and beginning of a new 24-h cycle [
133,
134,
135,
136,
137]. In the secondary loop, the CLOCK:BMAL1 heterodimer activates the transcription of
Rev-erbα/β genes, which belong to the orphan retinoic acid receptors family. In turn, REV-ERB proteins compete with ROR receptors for binding to ROR-response elements (RORE) sequences in the
Bmal1 promoter to repress or activate their transcription, respectively [
138,
139]. Also, the CLOCK:BMAL1 heterodimer regulates the expression of a set of genes known as clock-controlled genes through E-box sites in their promoter regions. In this way, the circadian clock exerts its control in molecular, biochemical, and physiologic processes, including cell cycle, proliferation, metabolism, senescence, and DNA repair, among others [
140,
141,
142,
143]. In addition, at the SCN, fluid communication between astrocytes and neurons was observed to coordinate and maintain circadian oscillations [
144].
5. Circadian Disruption and Its Implication in Cancer Biology
As postulated by Hanahan and Weinberg (2011), tumor cells share common features known as hallmarks of cancer that characterize how cancerous cells disrupt cellular homeostasis promoting tumor growth. These hallmarks include sustaining signaling promoting cellular proliferation, replicative immortality, the capability of evading cell death mechanisms, the capacity to avoid growth suppressors, the ability to trigger blood vessel formation (angiogenesis), the capacity of metastasis, the deregulation of cellular energetics, and the capability to evade the anti-tumoral immunological response. The features mentioned above are associated with crucial genomic instability and inflammation, contributing to tumor development [
145]. Several studies in the literature suggest tight crosstalk between the circadian clock function with tumorigenesis and cancer progression in different tumor models. It was evidenced that clock and clock-controlled genes regulate several pathways involved in cellular proliferation and growth under physiological conditions and that, when altered, may promote some of the hallmarks mentioned above of cancer, strongly suggesting that tumor cells can hijack the endogenous clock functioning to assure unrestricted proliferation, enhance the metabolism to supply their high energetics demands, and adapt and modify the microenvironment to promote tumor growth [
146,
147].
The molecular clock can positively or negatively modulate the different cell cycle phases. Moreover, several regulators implicated in cell cycle checkpoints show daily expression patterns [
148,
149,
150,
151]. Sustaining cell signaling is considered another hallmark of cancer, and several studies suggest its connection with the circadian clock, showing that proteins related to proliferation pathways exhibited circadian patterns of expression [
148,
152]. In this respect, Myc oncogenic activation is also observed when deregulation of sympathetic nervous system modulation of peripheral tissues occurs [
153]. Also, the circadian expression, stability, and activity of p53, one of the most studied tumor suppressors, is modulated by BMAL1 and PER2 [
154,
155,
156,
157,
158]. Lastly, the RAS/MAPK pathway was associated with alterations in the circadian clock, in which anomalous RAS activation impairs CLOCK:BMAL1 activity and up-regulates Ink4a/Arf [
159,
160].
Regarding tumor metabolism, cancer cells have high energetic demands to sustain exacerbated proliferation. Circadian disruption has been associated with changes in the cellular metabolic program, modulating glucose utilization, amino acid uptake, lipogenesis, glycerophospholipid metabolism, and β-oxidation [
128,
161,
162,
163]. This metabolic rearrangement is known as the Warburg effect, in which metabolism mainly occurs through glycolysis, as opposed to mitochondrial oxidative phosphorylation in normal cells. Also, this phenomenon is associated with a reduction in the tricarboxylic acid (TCA) cycle activity, an increase in the synthesis of fatty acids, and an enhanced NADPH formation. Remarkably, the circadian clock regulates NADPH levels, a critical anabolic intermediate that plays a crucial role in cancer development [
164]; it also regulates the expression of several genes involved in the transport and metabolism of glucose [
165,
166,
167].
Since the circadian clock plays an essential role in immune system regulation, alterations in clock function have been associated with aberrant inflammation, evasion of immunological surveillance, and immune cell functionality changes leading to cancer progression [
168,
169]. Lastly, cell death and DNA damage response are other mechanisms involved in the recognized hallmarks of cancer associated with the circadian clock deregulation [
147,
170,
171,
172,
173,
174,
175,
176,
177,
178] (
Figure 1).
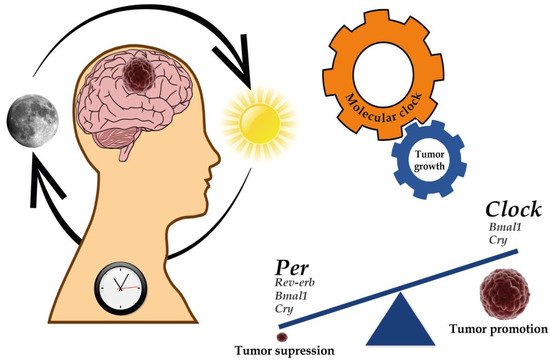
Figure 1. Implications of the circadian clock on cancer development and progression. Several genes of the molecular clock have been associated with both the regulation of GBM genesis and progression as well as tumor suppression. For further details, please see Table 3.
Table 3. Evidence of dysregulated clock gene expression in GBM.
| Gene |
Evidence of Gene Deregulation Associated with Gliomagenesis—Evidence of Potentiality for Therapeutic Targeting |
| Clock |
[209,211,213,219,223,224,225,226,227] |
| Bmal1 |
[21,22,161,209,219,220,221,222] |
| Per1 |
[21,161,213,225,227,228,229,230,231] |
| Per2 |
[213,227,228,230,231,232,233] |
| Per3 |
[213,225,227,231] |
| Cry1 |
[209,222,225,227,231,234] |
| Cry2 |
[213,222,225,231,234,235] |
| Npas2 |
[225,227] |
| Rev-erb |
[209,212,213,219,241,242] |
| RORα |
[213] |
| RORβ |
[213] |
| Timeless |
[227,243] |