 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tibor Pankotai | + 4655 word(s) | 4655 | 2021-08-10 12:03:44 | | | |
| 2 | Amina Yu | -1666 word(s) | 2989 | 2021-08-12 04:47:36 | | |
Video Upload Options
UV-induced DNA damage response and repair are extensively studied processes, as any malfunction in these pathways contributes to the activation of tumorigenesis. Although several proteins involved in these cellular mechanisms have been described, the entire repair cascade has remained unexplored. To identify new players in UV-induced repair, we performed a microarray screen, in which we found SerpinB10 (SPB10, Bomapin) as one of the most dramatically upregulated genes following UV irradiation. Here, we demonstrate that an increased mRNA level of SPB10 is a general cellular response following UV irradiation regardless of the cell type. We show that although SPB10 is implicated in the UV-induced cellular response, it has no indispensable function in cell survival upon UV irradiation. Nonetheless, we reveal that SPB10 might be involved in delaying the duration of DNA repair in interphase and also in S-phase cells. Additionally, we also highlight the interaction between SPB10 and H3. Based on our results, it seems that SPB10 protein is implicated in UV-induced stress as a “quality control protein”, presumably by slowing down the repair process.
1. Introduction
2. Results
2.1. Expression of several members of the SerpinB superfamily, including SPB10, is increased following UV irradiation
Environmental stress factors frequently induce DNA damage, which initiates the activation of DNA repair. These processes are tightly regulated, and post-translational regulation of the participating proteins is required for their proper function. Recently, we demonstrated by microarray experiment that UV irradiation contributes to the overexpression of certain members of the SerpinB family, including SerpinB2 (SPB2), SerpinB10 (SPB10), and SerpinB13 (SPB13), in Hker E6SFM cells, with 1.495-, 4.253-, and 1.180-fold increase (log2) upon UV irradiation, respectively (Figure S1).
To verify the microarray data, first we performed quantitative PCR (qPCR) on Hker E6SFM cells to measure the mRNA level of SPB10 in basal conditions and upon UV irradiation. We observed increased expression of SPB10 2 and 8 hours after UV irradiation compared to the control. The elevated mRNA level decreased 24 hours after the UV radiation; however, it remained 12.2 times higher compared to the control (Figure 1A).
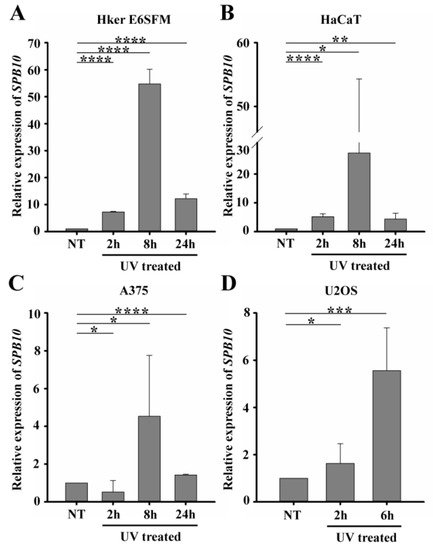
Figure 1. UV irradiation triggers elevation of SPB10 mRNA: Relative expression levels of SPB10 gene in (A) Hker E6SFM keratinocyte, (B) HaCaT keratinocyte, (C) A375 melanoma, and (D) U2OS osteosarcoma cells analyzed by qPCR. Data were normalized to 18S RNA and compared to control (NT). Means and standard deviations of three independent experimental triplicates are indicated as fold changes. Asterisks indicate statistical significance between datasets (t-test, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001).
To determine whether elevated gene expression of SPB10 could be observed in other cell lines as well, we performed qPCR experiments following UV irradiation for 2, 8, and 24 hours and with untreated samples in HaCaT keratinocyte and A375 melanoma cells. Similar to Hker E6SFM, we detected elevated SPB10 mRNA levels after UV irradiation in both cell lines (Figure 1B, C).
In order to reveal whether SPB10 plays a general role following UV radiation, we involved the U2OS osteosarcoma cell line in our study and performed the above-described experiment, since U2OS is generally used to examine DNA damage-induced cellular responses. We detected elevated SPB10 mRNA levels 2 and especially 6 hours after irradiation compared to the control samples in this cell line (Figure 1D). All of these results might indicate that the overexpression of SPB10 upon UV irradiation is not cell-line-specific, but a general cellular response.
2.2. SPB10 is dispensable for cell survival but influences DNA repair kinetics in interphase cells upon UV irradiation
Since our qPCR data suggested that SPB10 plays a role in UV-induced cellular responses, we examined whether it has any effect on cell survival in response to UV irradiation. To examine this hypothesis, we performed a viability assay on non-targeting scrambled siRNA (siSCR) and SPB10 siRNA-silenced U2OS cells and determined the ratio of live and dead cells 2, 4, 6, and 24 hours after UV radiation using Trypan blue staining (Figure 2A). As expected, we counted a reduced number of cells immediately after UV irradiation. Nonetheless, at later time points (24 hours after exposure), the amount of living cells increased, as by that point, the cells had been already recovered. However, we did not find any significant differences between the numbers of non-targeting (siSCR) and SPB10 siRNA-silenced cells, which indicates that SPB10 might have no effect on cell survival following UV irradiation.
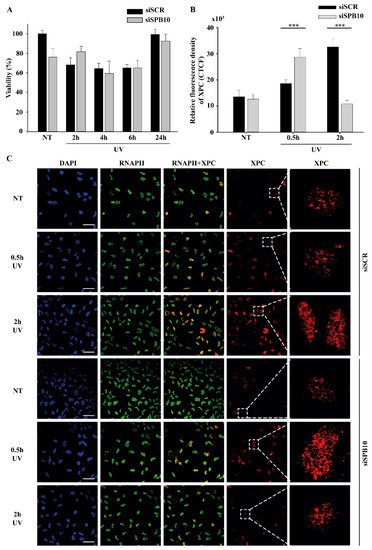
Figure 2. SPB10 is dispensable for cell survival but influences DNA repair kinetics in interphase cells upon UV irradiation: (A) Diagram represents cell viability (%) upon UV irradiation (2, 4, 6, and 24 hours) of non-targeting (siSCR) and SPB10 siRNA-silenced U2OS cells. (B) Quantification of relative fluorescence intensity (CTCF) of Xeroderma Pigmentosum C (XPC) protein detected on non-targeting (siSCR) and SPB10 siRNA-silenced interphase U2OS cells in basal conditions (NT) and following UV irradiation (0.5 and 2 hours). Asterisks indicate statistical significance between datasets (Mann–Whitney test, ***P ≤ 0.001). (C) Representative images of XPC protein (red) binding presumably to Nucleotide Excision Repair (NER) foci in non-targeting (siSCR, upstream) and SPB10 siRNA-silenced (siSPB10, downstream) U2OS cells in basal conditions (NT) and after UV irradiation (0.5 and 2 hours). Only the chromatin-bound proteins were visualized by CSK-immunocytochemistry. DAPI (blue) was used to visualize the nuclei, and RNA Polymerase II (RNAPII) (green) was used as control. For each condition, a higher magnification of a single cell is shown on the right side of the figure.
Although we did not find any changes in cell survival, showing that the SPB10 is not crucial for the UV-induced repair, it can still act as a fine-tune regulator protein in Nucleotide Excision Repair (NER). To test this, we monitored the repair kinetics of NER upon UV irradiation, by following the binding of the Xeroderma Pigmentosum C (XPC) protein presumably to the site of DNA damage by using CSK-immunocytochemistry of non-targeting (siSCR) and SPB10 siRNA-silenced U2OS cells. As we expected, the number of XPC foci was elevated after 30 minutes and 2 hours UV irradiation compared to the control sample in non-targeting (siSCR) U2OS cells. The repair kinetics was shifted in SPB10 siRNA-silenced cells 30 minutes following UV irradiation, while it was decreased 2 hours post-UV compared to its control (Figure 2B, C). Based on these results we assumed that SPB10 could influence the XPC binding, and the loss of SPB10 accelerates the repair of UV-induced damage.
2.3. SPB10 influences DNA repair kinetics in S-phase cells upon UV irradiation
Our data suggest that SPB10 may take part in regulating the repair of UV-induced DNA damage. UV-related T-T dimer formation also can results in replication block, which can be resolved by translesion (TLS) DNA polymerases, recruited by the proliferating cell nuclear antigen (PCNA) protein [18,34].
To reveal whether SPB10 is involved in this process, we first performed co-immunoprecipitation (co-IP) to study the association between SPB10 and PCNA. However, we did not detect interaction between these two proteins (data not shown).
Next, we tested whether SPB10 has any influence on the chromatin-bound PCNA recruitment upon UV irradiation. We made CSK-immunocytochemistry of non-targeting (siSCR) and SPB10 siRNA-silenced U2OS cells and we monitored the level of chromatin-bound PCNA in siSCR cells and 30 minutes after UV treatment. We observed less chromatin-bound PCNA upon UV irradiation in non-targeting (siSCR) siRNA treated U2OS cells, which might be caused by replication fork collapse mechanism. However, in the absence of SPB10, we could not observe this reduction in the PCNA level upon UV radiation (Figure 3A, B). According to these results, we hypothesized that SPB10 might involve in the fine-tuning of UV-induced repair mechanism also in S-phase cells.
To validate whether SPB10 indeed played a role in UV-related replication stress we performed post-replication repair (PRR) comet assay under physiological conditions and following UV irradiation of non-targeting (siSCR) and SPB10 siRNA-silenced U2OS cells and examined the presence of single-stranded DNA fragments generated by incomplete replication or UV light in S-phase cells following 0, 6, and 24 hours of UV treatment. As expected, after UV irradiation, damaged DNA strands (the “tail” of the comet) were accumulated in non-targeting (siSCR) cells, but only a limited amount of DNA damage was detected in SPB10 siRNA-silenced S-phase cells. Additionally, we observed faster repair, indicated by significantly shorter comet tails in the SPB10 siRNA-silenced cells (Figure 3 C, D). According to these results, our assumption is that in replicating cells, SPB10 is presumably involved in delaying the DNA repair process, thus facilitating a more precise repair mechanism to eliminate DNA damage.
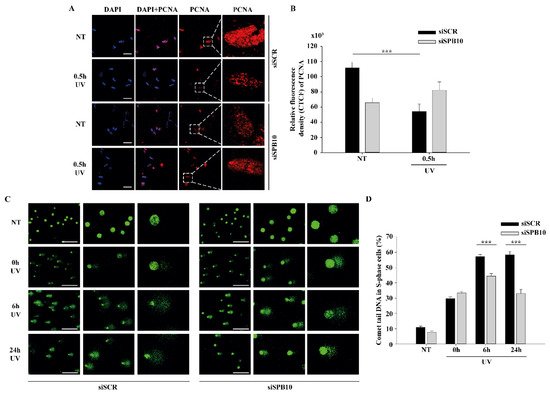
Figure 3 SPB10 influences DNA repair kinetics in S-phase cells upon UV irradiation: (A) Representative images of PCNA protein (red) binding in non-targeting (siSCR, upstream) and SPB10 siRNA-silenced (siSPB10, downstream) U2OS cells in basal conditions (NT) and after UV irradiation (0.5 hour). Only the chromatin-bound proteins were visualized by CSK-immunocytochemistry. DAPI (blue) was used to visualize the nuclei. For each condition, a higher magnification of a single cell is shown on the right side of the figure. (B) Quantification of relative fluorescence intensity (CTCF) based on measured foci number of proliferating cell nuclear antigen (PCNA) protein detected of non-targeting (siSCR) and SPB10 siRNA-silenced U2OS cells in basal conditions (NT) and following UV irradiation (0.5 hour). Asterisks indicate statistical significance between datasets (Mann–Whitney test, ***P ≤ 0.001). (C) Representative images of comet tail formation in non-targeting (siSCR, left) and SPB10 siRNA-silenced (siSPB10, right) bromodeoxyuridine (BrdU)-labelled S-phase U2OS cells in basal conditions (NT) and after UV irradiation (0, 6, and 24 hours). (D) Quantification of comet DNA tails detected on non-targeting (siSCR) and SPB10 siRNA-silenced S-phase U2OS cells in basal conditions and following UV irradiation (0, 6, and 24 hours). Asterisks indicate statistical significance between datasets (Mann–Whitney test, ***P ≤ 0.001).
2.4. SPB10 is associated with chromatin
The above-described data suggest that SPB10 might have an effect in the early steps of the UV-induced repair processes. To investigate whether this function takes place on the chromatin, we tested a possible association of SPB10 with the chromatin-related histone H3 protein following UV irradiation by co-IP in U2OS cells. For this, we transiently expressed SPB10-GFP fusion protein in untreated and UV-irradiated (2 and 4 hours) U2OS cells and performed an IP experiment using GFP antibody. Immunoprecipitated samples were analyzed by Western blot in order to detect co-immunoprecipitated H3 protein. We found that regardless of the cellular conditions, SPB10 showed interaction with H3 histones (Figure 4A and Figure S2A). A verifying reciprocal co-IP experiment using H3 specific antibody confirmed the interaction between H3 and SPB10 (Figure 4C and Figure S2B).
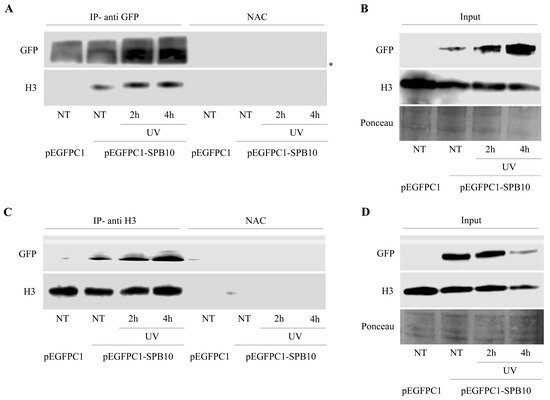
Figure 4. SPB10 shows interaction with H3: Immunoblot detection to reveal interaction between SPB10 and H3 in U2OS cells. Efficiency of immunoprecipitation experiment was controlled with (A) anti-GFP antibody (since cells were transfected with plasmid encoding SPB10-GFP fusion protein) and (C) anti-H3
3. Discussion
References
- Epstein, J.H. Photocarcinogenesis, skin cancer, and aging. J. Am. Acad. Dermatol. 1983, 9, 487–502.
- de Gruijl, F.R.; van Kranen, H.J.; Mullenders, L.H. UV-induced DNA damage, repair, mutations and oncogenic pathways in skin cancer. J. Photochem. Photobiol. B 2001, 63, 19–27.
- Kielbassa, C.; Roza, L.; Epe, B. Wavelength dependence of oxidative DNA damage induced by UV and visible light. Carcinogenesis 1997, 18, 811–816.
- Rünger, T.M.; Kappes, U.P. Mechanisms of mutation formation with long-wave ultraviolet light (UVA). Photodermatol. Photoimmunol. Photomed. 2008, 24, 2–10.
- de Gruijl, F.R. Photocarcinogenesis: UVA vs UVB. Methods Enzymol. 2000, 319, 359–366.
- Kabuyama, Y.; Homma, M.K.; Kurosaki, T.; Homma, Y. Early signaling events induced by 280-nm UV irradiation. Eur. J. Biochem. 2002, 269, 664–670.
- Pustovalova, Y.; MacIejewski, M.W.; Korzhnev, D.M. NMR mapping of PCNA interaction with translesion synthesis DNA polymerase Rev1 mediated by Rev1-BRCT domain. J. Mol. Biol. 2013, 425, 3091–3105.
- Waters, L.S.; Minesinger, B.K.; Wiltrout, M.E.; D’Souza, S.; Woodruff, R.V.; Walker, G.C. Eukaryotic Translesion Polymerases and Their Roles and Regulation in DNA Damage Tolerance. Microbiol. Mol. Biol. Rev. 2009, 73, 134–154.
- Guo, C.; Kosarek-Stancel, J.N.; Tang, T.S.; Friedberg, E.C. Y-family DNA polymerases in mammalian cells. Cell. Mol. Life Sci. 2009, 66, 2363–2381.
- Hoege, C.; Pfander, B.; Moldovan, G.-L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141.
- Moldovan, G.L.; Pfander, B.; Jentsch, S. PCNA, the Maestro of the Replication Fork. Cell 2007, 129, 665–679.
- Rizzo, A.A.; Korzhnev, D.M. The Rev1-Polζ translesion synthesis mutasome: Structure, interactions and inhibition. In Enzymes; Academic Press: Cambridge, MA, USA, 2019; Volume 45, pp. 139–181. ISBN 9780128173961.
- Acharya, N.; Patel, S.K.; Sahu, S.R.; Kumari, P. “PIPs” in DNA polymerase: PCNA interaction affairs. Biochem. Soc. Trans. 2020, 48, 2811–2822.
- Friedberg, E.C.; Lehmann, A.R.; Fuchs, R.P.P. Trading Places: How do DNA polymerases switch during translesion DNA synthesis? Mol. Cell 2005, 18, 499–505.
- Zhuang, Z.; Johnson, R.E.; Haracska, L.; Prakash, L.; Prakash, S.; Benkovic, S.J. Regulation of polymerase exchange between Polη and Polδ by monoubiquitination of PCNA and the movement of DNA polymerase holoenzyme. Proc. Natl. Acad. Sci. USA 2008, 105, 5361–5366.
- Maiorano, D.; El Etri, J.; Franchet, C.; Hoffmann, J.-S. Translesion Synthesis or Repair by Specialized DNA Polymerases Limits Excessive Genomic Instability upon Replication Stress. Int. J. Mol. Sci. 2021, 22, 3924.
- Guilliam, T.A.; Yeeles, J.T.P. Reconstitution of translesion synthesis reveals a mechanism of eukaryotic DNA replication restart. Nat. Struct. Mol. Biol. 2020, 27, 450–460.
- Cortez, D. Replication-Coupled DNA Repair. Mol. Cell 2019, 74, 866–876.
- Ujfaludi, Z.; Tuzesi, A.; Majoros, H.; Rothler, B.; Pankotai, T.; Boros, I.M. Coordinated activation of a cluster of MMP genes in response to UVB radiation. Sci. Rep. 2018, 8, 2660.
- Majoros, H.; Ujfaludi, Z.; Borsos, B.N.; Hudacsek, V.V.; Nagy, Z.; Coin, F.; Buzas, K.; Kovács, I.; Bíró, T.; Boros, I.M.; et al. SerpinB2 is involved in cellular response upon UV irradiation. Sci. Rep. 2019, 9, 2753.
- Polyanka, H.; Szabo, K.; Tax, G.; Tubak, V.; Kusz, E.; Ujfaludi, Z.; Boros, I.; Bata-Csorgo, Z.; Kemény, L.; Szell, M. Primary characterization of a novel HPV-E6 oncogene immortalized keratinocyte cell line. J. Investig. Dermatol. 2018, 131, 70.
- Szlavicz, E.; Szabo, K.; Groma, G.; Bata-Csorgo, Z.; Pagani, F.; Kemeny, L.; Szell, M. Splicing factors differentially expressed in psoriasis alter mRNA maturation of disease-associated EDA+ fibronectin. Mol. Cell. Biochem. 2017, 436, 189–199.
- Silverman, G.A.; Whisstock, J.C.; Askew, D.J.; Pak, S.C.; Luke, C.J.; Cataltepe, S.; Irving, J.A.; Bird, P.I. Human clade B serpins (ov-serpins) belong to a cohort of evolutionarily dispersed intracellular proteinase inhibitor clades that protect cells from promiscuous proteolysis. Cell. Mol. Life Sci. 2004, 61, 301–325.
- Baker, M.S.; Bleakley, P.; Woodrow, G.C.; Doe, W.F. Inhibition of cancer cell urokinase plasminogen activator by its specific inhibitor PAI-2 and subsequent effects on extracellular matrix degradation. Cancer Res. 1990, 50, 4676–4684.
- Sheng, S.; Truong, B.; Fredrickson, D.; Wu, R.; Pardee, A.B.; Sager, R. Tissue-type plasminogen activator is a target of the tumor suppressor gene maspin. Proc. Natl. Acad. Sci. USA 1998, 95, 499–504.
- Zhang, P.; Li, X.; He, Q.; Zhang, L.; Song, K.; Yang, X.; He, Q.; Wang, Y.; Hong, X.; Ma, J.; et al. TRIM21-SERPINB5 AIDS GMPS repression to protect nasopharyngeal carcinoma cells from radiation-induced apoptosis. J. Biomed. Sci. 2020, 27, 30.
- Mo, Y.; Zhang, K.; Feng, Y.; Yi, L.; Liang, Y.; Wu, W.; Zhao, J.; Zhang, Z.; Xu, Y.; Hu, Q.; et al. Epithelial serpinb10, a novel marker of airway eosinophilia in asthma, contributes to allergic airway inflammation. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2019, 316, L245–L254.
- Ahn, J.W.; Atwell, B.J.; Roberts, T.H. Serpin genes AtSRP2 and AtSRP3 are required for normal growth sensitivity to a DNA alkylating agent in Arabidopsis. BMC Plant Biol. 2009, 9, 52.
- Hsieh, H.-H.; Chen, Y.-C.; Jhan, J.-R.; Lin, J.-J. The serine protease inhibitor serpinB2 binds and stabilizes p21 in senescent cells. J. Cell Sci. 2017, 130, 3272–3281.
- Jiang, J.; Ding, Y.; Wu, M.; Chen, Y.; Lyu, X.; Lu, J.; Wang, H.; Teng, L. Integrated genomic analysis identifies a genetic mutation model predicting response to immune checkpoint inhibitors in melanoma. Cancer Med. 2020, 9, 8498–8518.
- Przygodzka, P.; Ramstedt, B.; Tengel, T.; Larsson, G.; Wilczynska, M. Bomapin is a redox-sensitive nuclear serpin that affects responsiveness of myeloid progenitor cells to growth environment. BMC Cell Biol. 2010, 11, 30.
- Chou, R.-H.; Wen, H.-C.; Liang, W.-G.; Lin, S.-C.; Yuan, H.-W.; Wu, C.-W.; Chang, W.-S.W. Suppression of the invasion and migration of cancer cells by SERPINB family genes and their derived peptides. Oncol. Rep. 2011, 27, 238–245.
- Shioji, G.; Ezura, Y.; Nakajima, T.; Ohgaki, K.; Fujiwara, H.; Kubota, Y.; Ichikawa, T.; Inoue, K.; Shuin, T.; Habuchi, T.; et al. Nucleotide variations in genes encoding plasminogen activator inhibitor-2 and serine proteinase inhibitor B10 associated with prostate cancer. J. Hum. Genet. 2005, 50, 507–515.
- Garmyn, M.; Young, A.R.; Miller, S.A. Mechanisms of and variables affecting UVR photoadaptation in human skin. Photochem. Photobiol. Sci. 2018, 17, 1932–1940.
- López-Camarillo, C.; Aréchaga Ocampo, E.; López Casamichana, M.; Pérez-Plasencia, C.; Álvarez-Sánchez, E.; Marchat, L.A. Protein Kinases and Transcription Factors Activation in Response to UV-Radiation of Skin: Implications for Carcinogenesis. Int. J. Mol. Sci. 2011, 13, 142–172.
- Kirillova, I.; Chaisson, M.; Fausto, N. Tumor necrosis factor induces DNA replication in hepatic cells through nuclear factor κB activation. Cell Growth Differ. 1999, 10, 819–828.
- Schleef, R.R.; Chuang, T.L. Protease Inhibitor 10 Inhibits Tumor Necrosis Factor α-induced Cell Death. J. Biol. Chem. 2000, 275, 26385–26389.
- Kojima, Y.; Machida, Y.; Palani, S.; Caulfield, T.R.; Radisky, E.S.; Kaufmann, S.H.; Machida, Y.J. FAM111A protects replication forks from protein obstacles via its trypsin-like domain. Nat. Commun. 2020, 11, 1318.
- Borsos, B.N.; Majoros, H.; Pankotai, T. Ubiquitylation-Mediated Fine-Tuning of DNA Double-Strand Break Repair. Cancers 2020, 12, 1617.
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. RNF168 Binds and Amplifies Ubiquitin Conjugates on Damaged Chromosomes to Allow Accumulation of Repair Proteins. Cell 2009, 136, 435–446.
- Stewart, G.S.; Panier, S.; Townsend, K.; Al-Hakim, A.K.; Kolas, N.K.; Miller, E.S.; Nakada, S.; Ylanko, J.; Olivarius, S.; Mendez, M.; et al. The RIDDLE Syndrome Protein Mediates a Ubiquitin-Dependent Signaling Cascade at Sites of DNA Damage. Cell 2009, 136, 420–434.
- Mattiroli, F.; Vissers, J.H.A.; Van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 2012, 150, 1182–1195.
- Gatti, M.; Pinato, S.; Maspero, E.; Soffientini, P.; Polo, S.; Penengo, L. A novel ubiquitin mark at the N-terminal tail of histone H2As targeted by RNF168 ubiquitin ligase. Cell Cycle 2012, 11, 2538–2544.


