Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Isabelle Moulonguet | + 2858 word(s) | 2858 | 2021-08-02 06:00:00 | | | |
| 2 | Rita Xu | -4 word(s) | 2854 | 2021-08-11 10:45:20 | | | | |
| 3 | Conner Chen | Meta information modification | 2854 | 2021-09-22 04:37:46 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Moulonguet, I. Panniculitis in Children. Encyclopedia. Available online: https://encyclopedia.pub/entry/13047 (accessed on 08 February 2026).
Moulonguet I. Panniculitis in Children. Encyclopedia. Available at: https://encyclopedia.pub/entry/13047. Accessed February 08, 2026.
Moulonguet, Isabelle. "Panniculitis in Children" Encyclopedia, https://encyclopedia.pub/entry/13047 (accessed February 08, 2026).
Moulonguet, I. (2021, August 11). Panniculitis in Children. In Encyclopedia. https://encyclopedia.pub/entry/13047
Moulonguet, Isabelle. "Panniculitis in Children." Encyclopedia. Web. 11 August, 2021.
Copy Citation
Panniculitides comprise a heterogenous group of inflammatory diseases that involve the subcutaneous adipose tissue. In children, these disorders are rare but can be difficult to diagnose.
panniculitis
histopathology
autoinflammatory diseases
1. Introduction
Panniculitides comprise a heterogenous group of inflammatory diseases that involve the subcutaneous adipose tissue. In children, these disorders are rare but can be difficult to diagnose. “Adult” types of panniculitis that occasionally occur in children will only be cited or just briefly described. Certain subtypes of panniculitis (such as Rothmann-Makai syndrome, Weber-Christian disease, and cytophagic histiocytic panniculitis) are no longer considered to be specific entities. Furthermore, genetic lipodystrophies characterised by the loss of adipose tissue at some body sites will not be described here.
Most types of panniculitis have the same clinical appearance (regardless of the aetiology), with tender, erythematous subcutaneous nodules that occur mostly where fatty tissue is prominent (i.e., on the legs, thighs, buttocks, and cheeks) [1][2]. The histopathologic assessment of panniculitis is often difficult and requires adequate tissue samples—including subcutaneous fat. As non-specific histopathologic findings are common for late panniculitis lesions, it is necessary to sample early lesions [3]. With an appropriate biopsy specimen and an adequate clinical-pathological correlation, a specific diagnosis can be made in most cases of panniculitis [4].
2. Specific Paediatric Panniculitides
2.1. Subcutaneous Fat Necrosis of the Newborn (SFN)
SFN is a rare variant of lobular panniculitis that appears in term or post-term newborns during the first days of life. The lesions are characterised clinically by indurated plaques and subcutaneous nodules that are primarily located on the buttocks, shoulders, cheeks, and thighs (Figure 1A). In some cases, the nodules and plaques can ulcerate and exude purulent material [5][6][7] (Figure 1B). The cause of SFN is unknown, although many cases are associated with perinatal asphyxia and meconium aspiration. The pathogenesis of SFN might be related to a greater saturated/unsaturated fatty acid ratio in the newborn, since saturated fatty acids have a greater tendency to crystallise in adipose tissue. SFN has an excellent prognosis, and spontaneous resolution within several weeks is common. However, delayed-onset hypercalcemia may occur, and so prolonged calcium monitoring is advisable. A biopsy is not always necessary because the clinical diagnosis is often straightforward. However, a biopsy can be useful in clinically misleading cases (such as multinodular cases, in particular) because the histological findings are specific.
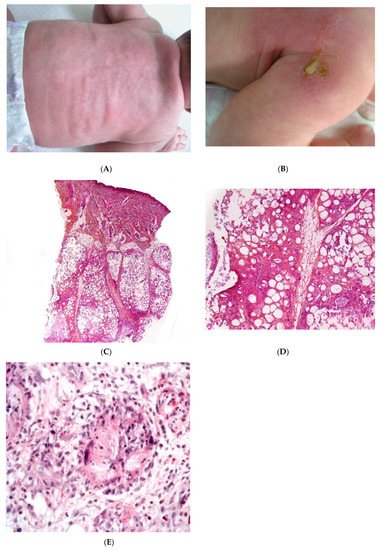
Figure 1. Subcutaneous fat necrosis of the newborn. (A) Indurated plaques on the neck and on the back of a newborn. (B) Significant purulent material of the shoulder Photo Dermatology Department Hopital Necker Enfants Malades. Paris France. (C) Lobular panniculitis without involvement of the dermis (original magnification ×20) (D) Many adipocytes are replaced by cells with finely eosinophilic granular cytoplasm that contain narrow needle-shaped clefts (original magnification ×100). (E) Needle-shaped crystals in radial fashion surrounded by histiocytes (original magnification ×400).
2.1.1. Histopathology
Most cases of SFN show characteristic features and are immediately recognisable as lobular panniculitis with necrosis of the fat lobule. It is a purely panniculitis, and the dermis is never affected. A high proportion of the adipocytes are replaced by cells with a finely eosinophilic, granular cytoplasm that contains radially arranged, narrow, needle-shaped clefts (Figure 1C–E). These clefts are characteristic of SFN and correspond to fatty acid crystals that have dissolved during sample processing. The adiponecrosis is associated with infiltration by inflammatory cells (lymphocytes, lipophages, and multinucleated giant cells), which can also contain these. Eosinophils and neutrophils can also be present. The neutrophil-rich variant of SFN can be difficult to distinguish from an infection [8]. Late-stage lesions can show septal fibrosis and areas of calcification within the fat lobule.
2.1.2. Differential Diagnoses
Intracytoplasmic, needle-shaped fatty acid crystals are quite characteristic of SFN but can be also observed in post-steroid panniculitis and in “sclerema neonatorum” (SN). This very rare disorder manifests itself within a few days of birth in premature newborns, with comorbidities such as congenital heart disease and other major developmental defects [9]. From a histopathologic point of view, it is notorious that, despite striking clinical features, histopathology usually minimal changes. In the subcutaneous fat, the inflammatory infiltrate is sparse or even absent [2]. Given that (i) no new cases of SN have been reported in the last few years and (ii) SFN and SN have clinical and pathological similarities, they are now considered to be variants of the same disease. Hence, SN may be a severe form of SFN that primarily manifests itself in premature newborns [2].
2.2. Post-Steroid Panniculitis
Post-steroid panniculitis is a very rare form of lobular panniculitis that has only been observed in children in whom systemic treatment with high-dose corticosteroids has been suddenly withdrawn. One to ten days after this withdrawal, erythematous subcutaneous nodules measuring 0.5 to 4 cm appear on the cheeks, the arms, or trunk (Figure 2A). The nodules may ulcerate, with scarring. The sudden withdrawal of corticosteroids might cause an increase in the saturated:unsaturated fatty acid ratio, and thus, crystal formation.
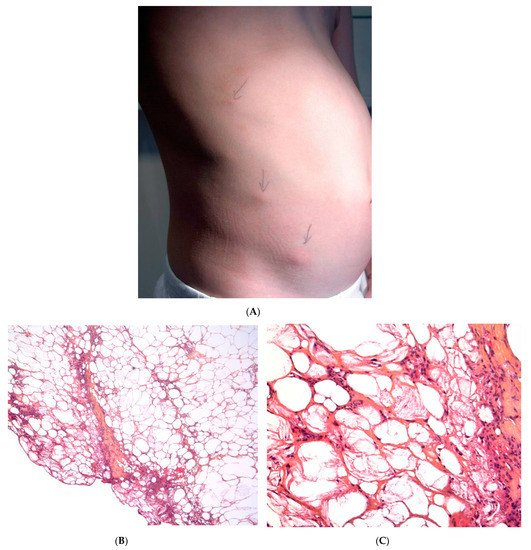
Figure 2. Post-steroid panniculitis. (A) Clinical presentation: subcutaneous nodules with overlying erythema. (B) Histopathologic features of post-steroid panniculitis: mostly lobular panniculitis with an inflammatory infiltrate of foamy histiocytes involving the fat lobules (original magnification ×40). (C) Needle-shaped clefts (original magnification ×400).
Histopathology
The findings are similar to SFN: a mostly lobular panniculitis features an inflammatory infiltrate of foamy histiocytes, with lymphocytes in the fat lobules. Some of the histiocytes show needle-shaped clefts in the cytoplasm (Figure 2B,C). In most cases, the inflammation is less intense and the crystals are less numerous than in SFN [10].
2.3. Cold Panniculitis
Cold panniculitis is related to cold exposure. It is more frequent in infants and children than in adults—probably because of age-related differences in fat composition. Cold panniculitis usually affects the cheeks and chin and appear 48 to 72 h after cold exposure. In particular, cold panniculitis is reported in infants after the application of ice bags for the treatment of supraventricular tachycardia [11] and as “popsicle panniculitis” in children sucking ice cubes or popsicles.
The clinic features include firm, indurated erythematous nodules or plaques with ill-defined margins on the cheeks and the submental region. The prognosis is excellent, and the disorder resolves spontaneously over a period of weeks to months. A biopsy is not usually required if the history of a cold exposure is reported. However, distinguishing clearly between SFN and cold panniculitis is difficult in some cases [2]. Given that SFN requires prolonged calcium monitoring, a biopsy may be needed if direct exposure to cold has not been reported.
Histopathology
Cold panniculitis is a mostly lobular form of panniculitis. The infiltrate mostly comprises lymphocytes and histiocytes in the fat lobules. The inflammation can affect both the interlobular septa and lobules, and is most intense at the dermal-subcutaneous junction. The dermis usually shows both superficial and deep perivascular infiltrates composed mostly of lymphocytes, in the absence of vasculitis.
2.4. Autoinflammatory Diseases
Early-onset panniculitis with systemic inflammation had been reported in cases of autoinflammatory diseases with or without an associated immunodeficiency [12][13]. Most cases have variable, non-specific histopathologic features: lobular or septal panniculitis with a mixture of cells or with a predominance of neutrophils or lymphocytes (Figure 3 and Figure 4). However, the histopathologic features can sometimes have diagnostic value, such as the granulomatous infiltrate characteristic of Blau syndrome (BS) or the polyarteritis-nodosa-like vasculitis in deficiency of adenosine deaminase 2 (DADA2). The presence of early-onset panniculitis with systemic inflammation should prompt the physician to screen for autoinflammatory disorders.
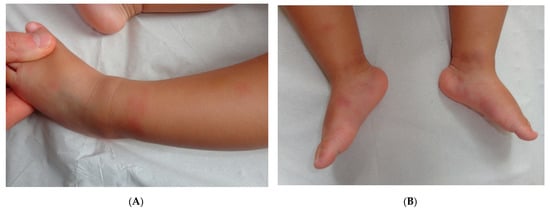
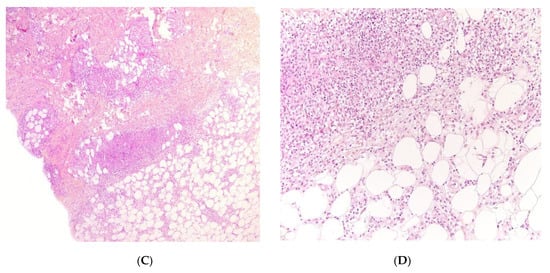
Figure 3. Panniculitis associated with inherited immunodeficiency. (A,B) Clinical presentation: Erythematous nodules of the leg of a 20 month-old boy Photo Dermatology Department Hopital Necker Enfants Malades. Paris France (C,D) Histopathology: inflammation of the deep dermis and the subcutis localised in the septa and the lobules composed of mixed infiltrate. (C) Original magnification ×80, (D) original magnification ×200.
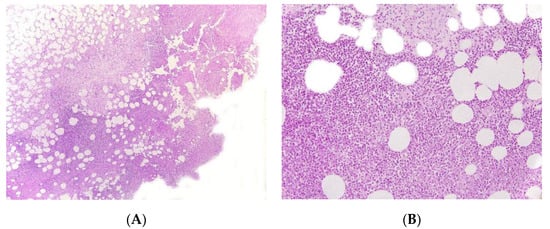
Figure 4. Panniculitis associated with inherited immunodeficiency. (A) Lobular panniculitis with dense neutrophilic infiltrate replacing part of the subcutis (original magnification ×80). (B) Closer view of showing entrapped adipocytes within the neutrophilic infiltrate (original magnification ×200).
2.4.1. Autoinflammatory Diseases in the Absence of Immunodeficiency
Proteasome-Associated Autoinflammatory Syndromes (PRAAS)
PRAAS (OMIM 256040) are a group of distinct clinical entities that have recently been recognised to share a common molecular cause. They include joint contractures, muscle atrophy, microcytic anemia and panniculitis-induced lipodystrophy syndrome (JMP), Nakajo-Nishimura syndrome (NNS, also referred to as Japanese autoinflammatory syndrome with lipodystrophy, JASL), and chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome (CANDLE). All these syndromes are characterised by the early onset of nodular, pernio-like, violaceous skin lesions with atypical neutrophil infiltrates, muscle atrophy, lipodystrophy, failure to thrive, and deformities of the hands and feet due to joint contractures. Recurrent periodic fever episodes and elevated acute phase reactant levels are usually present [14][15][16].
Histopathology
CANDLE is not a true panniculitis, since the reticular dermis and the hypodermis are affected and contain a perivascular and interstitial mononuclear inflammatory infiltrate. Many of the mononuclear cells exhibit large, vesicular, irregularly shaped nuclei, and thus, resemble atypical myeloid cells. Scattered mature neutrophils, some mature lymphocytes, and (in some cases) eosinophils are also present. Although leukocytoclasis is often present, there is no vasculitis [17]. Immunohistological studies have indicated that the cutaneous inflammatory infiltrate in CANDLE syndrome is polymorphous; it includes a mixture of immature myeloid cells that are strongly positive for myeloperoxidase, macrophages that are strongly positive for CD68 and CD163, and a moderate number of CD123-positive plasmacytoid dendritic cells.
Familial Mediterranean Fever
Familial Mediterranean fever classically consists of short, recurrent episodes of fever, serositis, arthritis, and erysipelas-like-erythema. The disorder results from mutations in the gene coding for pyrin (also known as marenostrin). Lobular panniculitis has been reported in adults but also in children, where tender, erythematous, bruise-like, warm, irregularly shaped nodules on the limbs and face coincide with the episodes of fever. The skin lesions heal as greyish macules without lipoatrophy. Panniculitis may be the main clinical manifestation, along with periods of fever [18].
Histopathology
Familial Mediterranean fever is a predominantly lobular neutrophilic panniculitis. Neither necrosis nor vasculitis is present [19].
Otulipenia
Otulipenia (also known as OTULIN-related autoinflammatory syndrome) is an autosomal recessive autoinflammatory disease caused by mutations in the FAM105B gene coding for OTU deubiquitinase with linear linkage specificity (OTULIN, a Met-1-specific deubiquitinase that downregulates the NF-kB signalling pathway). In clinical terms, patients present with early-onset, prolonged, recurrent episodes of fever, joint pain, abdominal pain, diarrhoea, and lymphadenopathy. A painful erythematous rash with nodules first noted in the neonatal period is the most frequent cutaneous manifestation.
Histopathology
Otulipenia is predominantly a septal form panniculitis with occasional vasculitis of small and medium-sized blood vessels [20].
BS
BS (also known as paediatric granulomatous arthritis) is usually caused by inherited dominant mutations in the NOD2/CARD15 gene. The disorder can also present sporadically as “early-onset sarcoidosis” after an acquired gene mutation. The skin, eyes, and joints are commonly involved, although some BS patients do not exhibit the full clinical triad. Skin involvement is most prominent and typically appears before joint symptoms and then eye involvement. Together with a histological analysis, a detailed clinical exploration of skin lesions by an expert dermatologist may enable the diagnosis of this orphan disease in early childhood. Early-onset skin lesions have a homogeneous, stereotypical clinical presentation as non-confluent erythematous or pigmented millimetre-size papules. In contrast, late-stage skin lesions have a more heterogeneous clinical presentation and may be wrongly diagnosed as erythema nodosum (EN), ichthyosiform dermatosis, livedoid lesions, or vasculitis [21]. Vouters et al. reported four children with infantile onset lobular panniculitis, high fever, uveitis, arthritis, and systemic granulomatous inflammation without CARD15 mutation [22].
Histopathology
BS is a granulomatous panniculitis, with an inflammatory infiltrate of typical, non-necrotising, non-caseating “sarcoid type” epithelioid and multinucleated giant cell granulomas.
Tumour Necrosis Factor Receptor-Associated Periodic Syndrome (TRAPS)
TRAPS is the most frequent autosomal dominant autoinflammatory disease. Mutations in the TNFRSF1A gene (coding for the tumour necrosis factor receptor 1) induce the overproduction of interleukin-1b. In children, variants of TRAPS usually occur as recurrent, irregular febrile episodes with generalised myalgia, joint pain, abdominal pain, ocular lesions, and (in about 80% of cases) skin involvement. The most frequent skin lesions are painful, migratory, centrifugal, tender, non-purpuric, well-demarcated erythematous plaques. Other manifestations include urticaria-like plaques, generalised serpiginous plaques, and small-vessel vasculitis [23][24].
Histopathology
TRAPS is mostly a lymphocytic, lobular form of panniculitis.
2.4.2. Autoinflammatory Syndromes with Inherited Immunodeficiency
Early-onset childhood panniculitis may reveal inherited immunodeficiency, and patients who present with unexplained panniculitis must undergo a detailed immunological screen because the clinical manifestations of immunodeficiency may not yet have emerged. An association with aseptic panniculitis was initially reported in infants with inherited immunodeficiency caused by mutations in the GATA2 gene (coding for a zinc finger transcription factor) or ADA2. Other mutations (in TRNT1, NFKB2, and LCK) have since been reported [12] (Figure 4, Figure 5 and Figure 6).
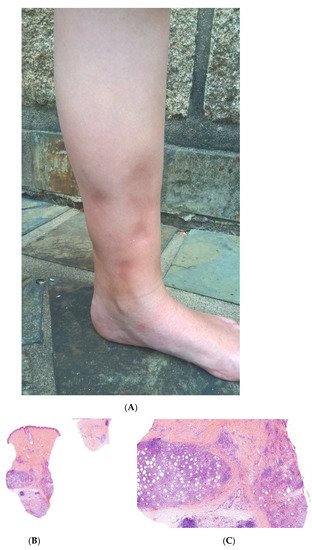
Figure 5. A Recurrent lipoatrophic panniculitis of children. (A) Areas of lipoatrophy in the leg Photo Dermatology Department Hopital Necker Enfants Malades. Paris France. (B,C) Lobular panniculitis with a mixed infiltrate, including neutrophils, as well as lymphocytes and macrophages. (B) Original magnification ×10, (C) original magnification ×40. Photo Courtesy of Christina Mittledorf Dermatologie, Venerologie und Allergologie Göttingen Germany.
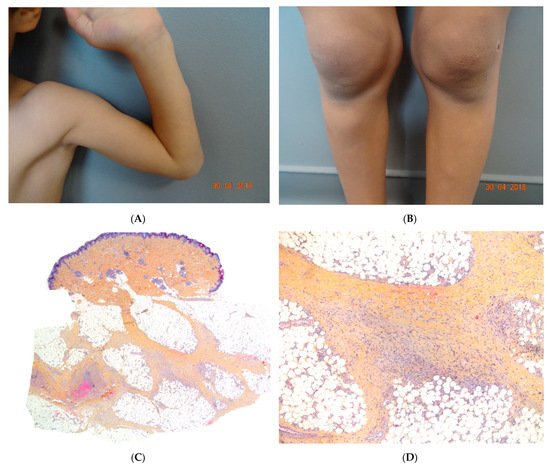
Figure 6. (A,B) Self-healing juvenile cutaneous mucinosis. Painful nodules of the limbs. Photo Courtesy of Christine Bodemer Dermatology Department Hopital Necker Enfants Malades. Paris France. (C,D) Chronic lobular panniculitis with features of proliferative fasciitis. (C) Original magnification ×20, (D) original magnification ×100.
Panniculitis in GATA2 Deficiency
The transcription factor GATA2 regulates haematopoietic differentiation, lymphatic development, and vascular development. Nearly 100 different GATA2 mutations have been described. Germline mutations arise spontaneously but are then transmitted via autosomal dominant inheritance. Patients with GATA2 mutations have very heterogeneous clinical presentations. The level of severity ranges from asymptomatic disease to life-threatening infections with respiratory failure and leukaemia. Up to 70% of patients with GATA2 mutations have dermatological features (mainly genital or extragenital warts) and up to a third have EN or panniculitis (usually on the lower limbs). These conditions can have several causes (nontuberculous mycobacterial infections, bacterial infections, or autoimmune phenomena) and may constitute the first manifestation of disease [25][26].
Histopathology
Panniculitis in GATA2 deficiency may be lobular or septal. Scleroderma-like changes deep in the subcutis (resembling deep morphoea) have been described [26].
Deficiency of Adenosine Deaminase 2 Deficiency
DADA2 is an autosomal recessive disorder caused by compound heterozygous missense mutations in ADA2. Skin manifestations may be the presenting symptoms of DADA2. They often include widespread livedo reticularis (often of the racemosa subtype) involving all the limbs and (in some cases) extending to the abdomen or trunk. Other skin manifestations include nodules, EN–like lesions, purpura, leg ulcers, and Raynaud symptoms [27][28]. Any diagnosis of polyarteritis nodosa in a child should always exclude DADA2, since early treatment with an anti-TNF may prevent a stroke.
Histopathology
A biopsy of the skin nodules may have much the same features as polyarteritis nodosa, with fibrinoid necrotising vasculitis affecting the small arteries (in acute panniculitis) and the dermal-subcutaneous junction (at the subacute or reparative stage). Other cases have been characterised by thrombosis of dermal and/or hypodermal capillaries, in the absence of vasculitis [28]. Overall, livedo racemosa with a large branching pattern, nodules or ulceration in a context of neurovascular events, recurrent fever, low immunoglobulin M levels, or paediatric onset is suggestive of DADA2.
2.5. Recurrent Lipoatrophic Panniculitis of Children
This term corresponds to a particular clinical presentation of idiopathic lobular panniculitis. It usually starts in childhood and is associated with fever, the systemic involvement of inflamed visceral fat, and (at the end of the inflammatory process) lipoatrophy. Other terms (such as “connective tissue panniculitis”, “lipophagic panniculitis”, and “annular lipoatrophy of the ankles”) have been used to describe cases with similar features. Use of the term “connective tissue panniculitis” is unfortunate because it may lead to confusion with other typical forms of panniculitis associated with connective tissue disorders (e.g., lupus panniculitis or panniculitis of dermatomyositis. Children with recurrent lipoatrophic panniculitis have inflamed, tender, subcutaneous nodules that appear mainly on the limbs (Figure 5A). In some cases, violaceous discoloration of the overlying skin is present. New lesions appear in subsequent attacks, with fever and slight malaise. Older lesions tend to subside and leave a striking, concentric, annular lipoatrophy.
Histopathology
Recurrent lipoatrophic panniculitis of children is a lobular form with a mixed but predominantly lymphocytic infiltrate that also contains histiocytes, eosinophils, plasma cells, and neutrophils (Figure 5B,C). The degree of lipoatrophy is variable. Lipidised giant cells are usually seen in the conditions called “lipophagic panniculitis of children” and “annular lipoatrophy of the ankles”. Several cases have been linked to autoimmune disorders: insulin-dependent diabetes mellitus, juvenile rheumatoid arthritis, Graves’ disease, Hashimoto thyroiditis, alopecia areata, vitiligo, coeliac disease, Raynaud’s phenomenon, Crohn’s disease, and partial IgA deficiency [29].
2.6. Panniculitis in Self-Healing Juvenile Cutaneous Mucinosis (SHJCM)
SHJCM typically presents as an acute eruption in an otherwise healthy child. The cutaneous lesions occur suddenly 3 to 10 days after prodromal fever and spread within a few days to become subcutaneous noninflammatory nodules on the head, hands, elbows, arms, and knees (Figure 6A,B). Additional nontender ivory-white papules may arise on the hands, head, and trunk at disease onset or during disease progression. Nodules are a common clinical feature and can precede the papules. Periorbital oedema, asthenia, and joint pain may also be encountered.
Histopathology
In the absence of mucinous papules, the diagnosis of SHJCM is challenging. A histopathological assessment of the papules reveals moderate dermal mucin deposition, whereas the nodules show features of proliferative fasciitis or non-specific chronic lobular panniculitis (Figure 6C) [30].
References
- Bodemer, C. Panniculitis in Harper’s Textbook of Pediatric Dermatology, 4th ed.; Wiley-Blackwell: Oxford, UK, 5 December 2019; pp. 1207–1220. (In English)
- Torrelo, A.; Hernández, A. Panniculitis in children. Dermatol. Clin. 2008, 26, 491–500.
- Polcari, I.C.; Stein, S.L. Panniculitis in childhood. Dermatol. Ther. 2010, 23, 356–367.
- Requena, L.; Yus, E.S. Panniculitis. Part I. Mostly septal panniculitis. J. Am. Acad. Dermatol. 2001, 45, 163–183.
- Del Pozzo-Magaña, B.R.; Ho, N. Subcutaneous Fat Necrosis of the Newborn: A 20-Year Retrospective Study. Pediatr. Dermatol. 2016, 33, e353–e355.
- Lara, L.G.; Villa, A.V.; Rivas, M.M.; Capella, M.S.; Prada, F.; Enseñat, M.A.G. Subcutaneous Fat Necrosis of the Newborn: Report of Five Cases. Pediatr. Neonatol. 2017, 58, 85–88.
- Stefanko, N.S.; Drolet, B.A. Subcutaneous fat necrosis of the newborn and associated hypercalcemia: A systematic review of the literature. Pediatr. Dermatol. 2019, 36, 24–30.
- Ricardo-Gonzalez, R.R.; Lin, J.R.; Mathes, E.F.; McCalmont, T.H.; Pincus, L.B. Neutrophil-rich subcutaneous fat necrosis of the newborn: A potential mimic of infection. J. Am. Acad. Dermatol. 2016, 75, 177–185.
- Zeb, A.; Darmstadt, G.L. Sclerema neonatorum: A review of nomenclature, clinical presentation, histological features, differential diagnoses and management. J. Perinatol. 2008, 28, 453–460.
- Silverman, R.A.; Newman, A.J.; LeVine, M.J.; Kaplan, B. Poststeroid panniculitis: A case report. Pediatr. Dermatol. 1988, 5, 92–93.
- Malia, L.; Wang, A.; Scheiner, L.; Laurich, V.M. Cold Panniculitis After Ice Therapy for Supraventricular Tachycardia. Pediatr. Emerg. Care 2019, 35, e174–e176.
- Bader-Meunier, B.; Rieux-Laucat, F.; Touzot, F.; Frémond, M.L.; André-Schmutz, I.; Fraitag, S.; Bodemer, C. Inherited Immunodeficiency: A New Association with Early-Onset Childhood Panniculitis. Pediatrics 2018, 141 (Suppl. 5), S496–S500.
- Escudier, A.; Mauvais, F.X.; Bastard, P.; Boussard, C.; Jaoui, A.; Koskas, V.; Lecoq, E.; Michel, A.; Orcel, M.C.; Truelle, P.E.; et al. Peau et fièvres récurrentes auto-inflammatoires [Dermatological features of auto-inflammatory recurrent fevers]. Arch. Pediatr. 2018, 25, 150–162. (In French)
- Torrelo, A.; Patel, S.; Colmenero, I.; Gurbindo, D.; Lendínez, F.; Hernández, A.; López-Robledillo, J.C.; Dadban, A.; Requena, L.; Paller, A.S. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. J. Am. Acad. Dermatol. 2010, 62, 489–495.
- Ohmura, K. Nakajo-Nishimura syndrome and related proteasome-associated autoinflammatory syndromes. J. Inflamm. Res. 2019, 12, 259–265.
- Volpi, S.; Picco, P.; Caorsi, R.; Candotti, F.; Gattorno, M. Type I interferonopathies in pediatric rheumatology. Pediatr. Rheumatol. Online J. 2016, 14, 35.
- Torrelo, A.; Colmenero, I.; Requena, L.; Paller, A.S.; Ramot, Y.; Richard Lee, C.C.; Vera, A.; Zlotogorski, A.; Goldbach-Mansky, R.; Kutzner, H. Histologic and Immunohistochemical Features of the Skin Lesions in CANDLE Syndrome. Am. J. Dermatopathol. 2015, 37, 517–522.
- Figueras-Nart, I.; Mascaró, J.M., Jr.; Solanich, X.; Hernández-Rodríguez, J. Dermatologic and Dermatopathologic Features of Monogenic Autoinflammatory Diseases. Front. Immunol. 2019, 10, 2448.
- Leiva-Salinas, M.; Betlloch, I.; Arribas, M.P.; Francés, L.; Pascual, J.C. Neutrophilic lobular panniculitis as an expression of a widened spectrum of familial mediterranean fever. JAMA Dermatol. 2014, 150, 213–214.
- Zhou, Q.; Yu, X.; Demirkaya, E.; Deuitch, N.; Stone, D.; Tsai, W.L.; Kuehn, H.S.; Wang, H.; Yang, D.; Park, Y.H.; et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc. Natl. Acad. Sci. USA 2016, 113, 10127–10132.
- Poline, J.; Fogel, O.; Pajot, C.; Miceli-Richard, C.; Rybojad, M.; Galeotti, C.; Grouteau, E.; Hachulla, E.; Brissaud, P.; Cantagrel, A.; et al. Early-onset granulomatous arthritis, uveitis and skin rash: Characterization of skin involvement in Blau syndrome. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 340–348.
- Wouters, C.H.; Martin, T.M.; Stichweh, D.; Punaro, M.; Doyle, T.M.; Lewis, J.A.; Quartier, P.; Rose, C.D. Infantile onset panniculitis with uveitis and systemic granulomatosis: A new clinicopathologic entity. J. Pediatr. 2007, 151, 707–709.
- Toro, J.R.; Aksentijevich, I.; Hull, K.; Dean, J.; Kastner, D.L. Tumor necrosis factor receptor-associated periodic syndrome: A novel syndrome with cutaneous manifestations. Arch. Dermatol. 2000, 136, 1487–1494.
- Cattalini, M.; Meini, A.; Monari, P.; Gualdi, G.; Arisi, M.; Pelucchi, F.; Bolognini, S.; Gattorno, M.; Calzavara-Pinton, P.G.; Plebani, A. Recurrent migratory angioedema as cutaneous manifestation in a familiar case of TRAPS: Dramatic response to Anakinra. Dermatol. Online J. 2013, 19, 20405.
- Polat, A.; Dinulescu, M.; Fraitag, S.; Nimubona, S.; Toutain, F.; Jouneau, S.; Poullot, E.; Droitcourt, C.; Dupuy, A. Skin manifestations among GATA2-deficient patients. Br. J. Dermatol. 2018, 178, 781–785.
- Spinner, M.A.; Sanchez, L.A.; Hsu, A.P.; Shaw, P.A.; Zerbe, C.S.; Calvo, K.R.; Arthur, D.C.; Gu, W.; Gould, C.M.; Brewer, C.C.; et al. GATA2 deficiency: A protean disorder of hematopoiesis, lymphatics, and immunity. Blood 2014, 123, 809–821.
- Chasset, F.; Fayand, A.; Moguelet, P.; Kouby, F.; Bonhomme, A.; Franck, N.; Goldman-Lévy, G.; Fraitag, S.; Barbaud, A.; Queyrel, V.; et al. Clinical and pathological dermatological features of deficiency of adenosine deaminase 2: A multicenter, retrospective, observational study. J. Am. Acad. Dermatol. 2020, 83, 1794–1798.
- Shwin, K.W.; Lee, C.R.; Goldbach-Mansky, R. Dermatologic Manifestations of Monogenic Autoinflammatory Diseases. Dermatol. Clin. 2017, 35, 21–38.
- Torrelo, A.; Noguera-Morel, L.; Hernández-Martín, A.; Clemente, D.; Barja, J.M.; Buzón, L.; Azorín, D.; de Jesús, A.A.; López-Robledillo, J.C.; Colmenero, I.; et al. Recurrent lipoatrophic panniculitis of children. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 536–543.
- Luchsinger, I.; Coulombe, J.; Rongioletti, F.; Haspeslagh, M.; Dompmartin, A.; Melki, I.; Dagher, R.; Bader-Meunier, B.; Fraitag, S.; Bodemer, C. Self-healing juvenile cutaneous mucinosis: Clinical and histopathologic findings of 9 patients: The relevance of long-term follow-up. J. Am. Acad. Dermatol. 2018, 78, 1164–1170.
More
Information
Subjects:
Anthropology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.0K
Revisions:
3 times
(View History)
Update Date:
22 Sep 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No