 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Don Davies | + 1560 word(s) | 1560 | 2021-07-29 09:54:03 |
Video Upload Options
Nuclear factor erythroid 2-related factor 2 (Nrf2) is an important transcription factor that reduces oxidative stress. When reactive oxygen species (ROS) or reactive nitrogen species (RNS) are detected, Nrf2 translocates from the cytoplasm into the nucleus and binds to the antioxidant response element (ARE), which regulates the expression of antioxidant and anti-inflammatory genes. Nrf2 impairments are observed in the majority of neurodegenerative disorders, including Alzheimer’s disease (AD). The classic hallmarks of AD include β-amyloid (Aβ) plaques, and neurofibrillary tangles (NFTs). Oxidative stress is observed early in AD and is a novel therapeutic target for the treatment of AD. The nuclear translocation of Nrf2 is impaired in AD compared to controls. Increased oxidative stress is associated with impaired memory and synaptic plasticity. The administration of Nrf2 activators reverses memory and synaptic plasticity impairments in rodent models of AD. Therefore, Nrf2 activators are a potential novel therapeutic for neurodegenerative disorders including AD.
1. Introduction
Oxidative stress is involved with the occurrence and progression of AD. Aβ elevation is associated with increased levels of oxidation products from proteins, lipids and nucleic acids in the hippocampus and cortex of humans with AD [1]. In contrast, lower Aβ levels in the brain are correlated with lower oxidative stress markers [2]. Aβ plaques can reduce Ca 2+ storage in the endoplasmic reticulum, which results in an excess of Ca 2+ in the cytosol [3]. Due to the excess of cytosolic Ca 2+ , glutathione (GSH) levels are decreased and reactive oxygen species (ROS) can accumulate in the neurons [4]. The oxidative stress in AD patients may be a result of excitotoxicity from the glutamatergic N -methyl- d -aspartate (NMDA) receptors. NMDA receptor activation in AD has been shown to result in an excessive influx of Ca 2+ by increasing cell permeability and generation of ROS and reactive nitrogen species [5][6]. In addition, Aβ can initiate free radical formation by activating NADPH oxidase [7]. Furthermore, abnormal aggregates composed of p-Tau protein lead to increased ROS production in AD. ROS was the key result of impaired axonal transport and caused by abnormal p-Tau protein [8].
Nrf2 is a key endogenous modulator in the protection against oxidative stress. In response to oxidative stress, Nrf2 translocates from the cytoplasm into the nucleus and activates genetic expression with antioxidant activity. AD patients had less nuclear Nrf2 in the CA1 region of their hippocampus than the controls despite oxidative stress markers in the hippocampal neurons of patients with AD [9]. This indicates that Nrf2 was not translocating from the cytoplasm into the nucleus in hippocampal neurons in patients with AD, despite oxidative stress markers in these neurons and an abundance of nuclear Nrf2 in the neurologically normal age matched controls (see Figure 1 ). Therefore, some process may be blocking Nrf2 nuclear activity, which may contribute to neuronal dysfunction. The levels of cytoplasmic Nrf2 are not different between age-matched controls and patients with AD. Albeit, the nuclear impairment is not the result of a general loss of Nrf2 protein but could reflect dysfunctional nuclear trafficking. Since the two hallmarks of AD are misfolded proteins, Aβ plaques and NFT, it is likely that endoplasmic reticulum stress is active in the hippocampus during the progression of AD, which may alter the Nrf2 pathway in the hippocampus. Methylene blue treatment in a mouse model of tauopathy increased the activation of Nrf2 and reduced tauopathy and oxidative stress [10]. Treatment with methylene blue was also associated with improved behavior with reduced locomotor abnormality, reduced anxiety abnormality, and improvement in learning and memory. Therefore, methylene blue may be a novel treatment option for people with AD because methylene blue reduces tau, which is one of the hallmarks of AD.
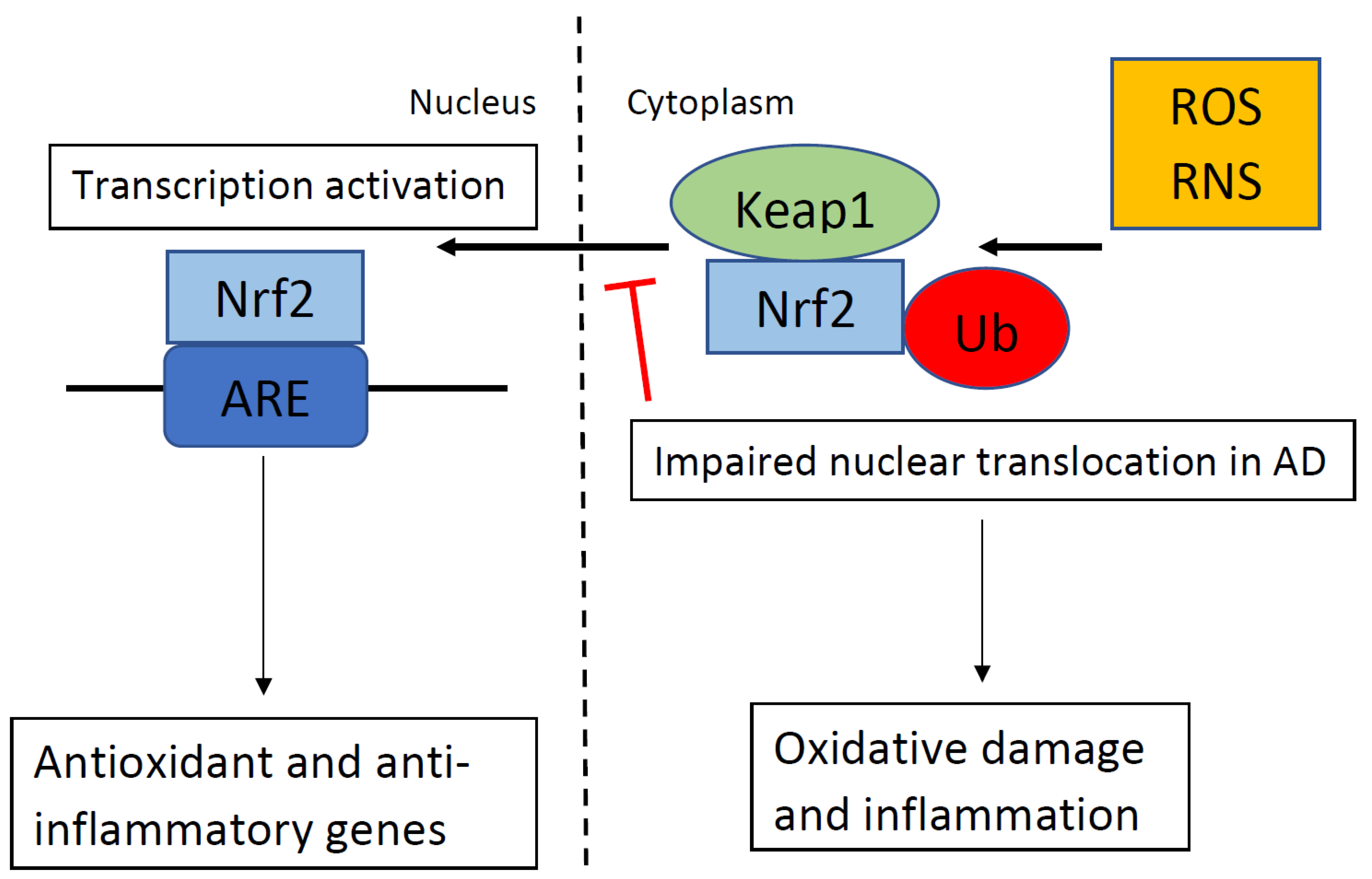 Figure 1. In the absence of oxidative stress. Kelch-like ECH-associating protein 1 (Keap1) suppresses the activation of nuclear factor erythroid 2-related factor 2 (Nrf2) and targets Nrf2 for ubiquitination (Ub). When Keap1 detects oxidative stress via reactive oxygen species (ROS) or reactive nitrogen species (RNS), as shown in the top right portion of the figure, Keap1 ends the inhibition of Nrf2, and Nrf2 translocates into the nucleus. Nrf2 binds to the antioxidant response element (ARE), which regulates the expression of antioxidant and anti-inflammatory genes. In Alzheimer’s disease (AD), nuclear translocation in response to ROS/RNS is impaired, which results in oxidative damage and inflammation.
Figure 1. In the absence of oxidative stress. Kelch-like ECH-associating protein 1 (Keap1) suppresses the activation of nuclear factor erythroid 2-related factor 2 (Nrf2) and targets Nrf2 for ubiquitination (Ub). When Keap1 detects oxidative stress via reactive oxygen species (ROS) or reactive nitrogen species (RNS), as shown in the top right portion of the figure, Keap1 ends the inhibition of Nrf2, and Nrf2 translocates into the nucleus. Nrf2 binds to the antioxidant response element (ARE), which regulates the expression of antioxidant and anti-inflammatory genes. In Alzheimer’s disease (AD), nuclear translocation in response to ROS/RNS is impaired, which results in oxidative damage and inflammation.
Initially, ROS was thought to only have negative physiological effects. However, others have observed the beneficial effects of ROS on mitochondria and in various cellular pathways [11][12]. Low levels of ROS are shown to have beneficial effects while high levels of ROS are associated with AD, suggesting a threshold determines whether ROS is beneficial or harmful [13]. The low levels of ROS regulate various cellular pathways, such as H2O2 regulating various signaling pathways with proteins containing cysteine residues [14]. Given the beneficial effects of low levels of ROS, Nrf2 activators should only be considered when ROS levels have crossed the threshold from beneficial into harmful.
2. Cross-Talk between the NF-ĸB and Nrf2 Signaling Pathways
Nrf2 signaling contributes to the anti-inflammatory process by regulating target genes via the antioxidant response element (ARE) and Keap1 system [15]. The Keap1/Nrf2/ARE signaling pathway mostly regulates the expression of anti-inflammatory genes and ultimately blocks the progression of inflammation [15]. Oxidative-stress mediated NF-κB activation can be blocked by the Keap1/Nrf2/ARE pathway [16][17]. NF-κB impacts the Keapl/Nrf2/ARE signaling pathway in three parts. First, Keap1 degrades IKK, which prevents the phosphorylation of NF-κB [16]. Second, oxidative stress, which activates IKK leads to phosphorylation of NF-κB and translocation of NF-κB from the cytoplasm into the nucleus stimulates the production of proinflammatory cytokines such as IL-1, IL-6, TNF-α, inducible nitric oxide synthase (iNOS), and cyclooxygenase-2 (COX-2) [18][19]. Ultimately, COX-2 reacts with Keap1 and activates Nrf2, which leads to the suppression of oxidative stress-mediated NF-κB activation [20]. Third, Nrf2 binds to CBP and other transcriptional machinery to begin ARE-driven gene transcription [21]. However, NF-κB inhibits Nrf2 activation by competing with Nrf2 for CBP and ultimately reducing ARE gene expression [21]. Overall, the Keap1/Nrf2/ARE signaling pathway inhibits the production of proinflammation [20]. Moreover, it has been demonstrated that Nrf2 directly regulates the expression of anti-inflammatory mediators such as CD36, IL-17D, macrophage receptor, and G protein-coupled receptor (GPCR) kinase, which suppresses the progression of inflammatory responses [22][23][24]. Nrf2 induces the anti-inflammatory phenotype of microglia and macrophages, while it decreases LPS-induced transcription of other NF-κB target genes [25][26]. Nrf2 increases cysteine and GSH levels in macrophages. However, depletion of GSH triggers macrophages to Nrf2 activation by LPS [27]. These findings show that Nrf2 acts as an anti-inflammatory marker, which is critical for regulating inflammatory responses.
Nrf2 and NF-κB Crosstalk with Other Transcription Factors
3. Conclusions
References
- Butterfield, D.A.; Lauderback, C.M. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic. Biol. Med. 2002, 32, 1050–1060.
- Hensley, K.; Hall, N.; Subramaniam, R.; Cole, P.; Harris, M.; Aksenov, M.; Aksenova, M.; Gabbita, S.P.; Wu, J.F.; Carney, J.M.; et al. Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J. Neurochem. 1995, 65, 2146–2156.
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxid. Med. Cell. Longev. 2017, 2017, 2525967.
- Ferreiro, E.; Oliveira, C.R.; Pereira, C.M. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol. Dis. 2008, 30, 331–342.
- Nakamura, T.; Lipton, S.A. Redox modulation by S-nitrosylation contributes to protein misfolding, mitochondrial dynamics, and neuronal synaptic damage in neurodegenerative diseases. Cell Death Differ. 2011, 18, 1478–1486.
- Nakamura, T.; Lipton, S.A. Preventing Ca2+-mediated nitrosative stress in neurodegenerative diseases: Possible pharmacological strategies. Cell Calcium 2010, 47, 190–197.
- Shelat, P.B.; Chalimoniuk, M.; Wang, J.H.; Strosznajder, J.B.; Lee, J.C.; Sun, A.Y.; Simonyi, A.; Sun, G.Y. Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J. Neurochem. 2008, 106, 45–55.
- Jiang, T.; Sun, Q.; Chen, S. Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog. Neurobiol. 2016, 147, 1–19.
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85.
- Stack, C.; Jainuddin, S.; Elipenahli, C.; Gerges, M.; Starkova, N.; Starkov, A.A.; Jove, M.; Portero-Otin, M.; Launay, N.; Pujol, A.; et al. Methylene blue upregulates Nrf2/ARE genes and prevents tau-related neurotoxicity. Hum. Mol. Genet. 2014, 23, 3716–3732.
- Gomez-Cabrera, M.C.; Salvador-Pascual, A.; Cabo, H.; Ferrando, B.; Vina, J. Redox modulation of mitochondriogenesis in exercise. Does antioxidant supplementation blunt the benefits of exercise training? Free Radic. Biol. Med. 2015, 86, 37–46.
- Santos, A.L.; Sinha, S.; Lindner, A.B. The Good, the Bad, and the Ugly of ROS: New Insights on Aging and Aging-Related Diseases from Eukaryotic and Prokaryotic Model Organisms. Oxid. Med. Cell. Longev. 2018, 2018, 1941285.
- Abate, G.; Vezzoli, M.; Sandri, M.; Rungratanawanich, W.; Memo, M.; Uberti, D. Mitochondria and cellular redox state on the route from ageing to Alzheimer’s disease. Mech. Ageing Dev. 2020, 192, 111385.
- Antunes, F.; Brito, P.M. Quantitative biology of hydrogen peroxide signaling. Redox Biol. 2017, 13, 1–7.
- Tu, W.; Wang, H.; Li, S.; Liu, Q.; Sha, H. The Anti-Inflammatory and Anti-Oxidant Mechanisms of the Keap1/Nrf2/ARE Signaling Pathway in Chronic Diseases. Aging Dis. 2019, 10, 637–651.
- Lee, D.F.; Kuo, H.P.; Liu, M.; Chou, C.K.; Xia, W.; Du, Y.; Shen, J.; Chen, C.T.; Huo, L.; Hsu, M.C.; et al. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol. Cell 2009, 36, 131–140.
- Bellezza, I.; Tucci, A.; Galli, F.; Grottelli, S.; Mierla, A.L.; Pilolli, F.; Minelli, A. Inhibition of NF-kappaB nuclear translocation via HO-1 activation underlies alpha-tocopheryl succinate toxicity. J. Nutr. Biochem. 2012, 23, 1583–1591.
- Karin, M.; Yamamoto, Y.; Wang, Q.M. The IKK NF-kappa B system: A treasure trove for drug development. Nat. Rev. Drug Discov. 2004, 3, 17–26.
- Ganesh Yerra, V.; Negi, G.; Sharma, S.S.; Kumar, A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-kappaB pathways in diabetic neuropathy. Redox Biol. 2013, 1, 394–397.
- Ahmed, S.M.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597.
- Liu, G.H.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta BBA Mol. Cell Res. 2008, 1783, 713–727.
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203.
- Saddawi-Konefka, R.; Seelige, R.; Gross, E.T.; Levy, E.; Searles, S.C.; Washington, A., Jr.; Santosa, E.K.; Liu, B.; O’Sullivan, T.E.; Harismendy, O.; et al. Nrf2 Induces IL-17D to Mediate Tumor and Virus Surveillance. Cell Rep. 2016, 16, 2348–2358.
- Ishii, T.; Itoh, K.; Ruiz, E.; Leake, D.S.; Unoki, H.; Yamamoto, M.; Mann, G.E. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: Activation by oxidatively modified LDL and 4-hydroxynonenal. Circ. Res. 2004, 94, 609–616.
- Rojo, A.I.; McBean, G.; Cindric, M.; Egea, J.; Lopez, M.G.; Rada, P.; Zarkovic, N.; Cuadrado, A. Redox control of microglial function: Molecular mechanisms and functional significance. Antioxid. Redox Signal. 2014, 21, 1766–1801.
- Brune, B.; Dehne, N.; Grossmann, N.; Jung, M.; Namgaladze, D.; Schmid, T.; von Knethen, A.; Weigert, A. Redox control of inflammation in macrophages. Antioxid. Redox Signal. 2013, 19, 595–637.
- Diotallevi, M.; Checconi, P.; Palamara, A.T.; Celestino, I.; Coppo, L.; Holmgren, A.; Abbas, K.; Peyrot, F.; Mengozzi, M.; Ghezzi, P. Glutathione Fine-Tunes the Innate Immune Response toward Antiviral Pathways in a Macrophage Cell Line Independently of Its Antioxidant Properties. Front. Immunol. 2017, 8, 1239.
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528.
- Toth, R.K.; Warfel, N.A. Strange Bedfellows: Nuclear Factor, Erythroid 2-Like 2 (Nrf2) and Hypoxia-Inducible Factor 1 (HIF-1) in Tumor Hypoxia. Antioxidants 2017, 6, 27.
- Kim, T.H.; Hur, E.G.; Kang, S.J.; Kim, J.A.; Thapa, D.; Lee, Y.M.; Ku, S.K.; Jung, Y.; Kwak, M.K. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1alpha. Cancer Res. 2011, 71, 2260–2275.
- Lu, Y.; Wang, B.; Shi, Q.; Wang, X.; Wang, D.; Zhu, L. Brusatol inhibits HIF-1 signaling pathway and suppresses glucose uptake under hypoxic conditions in HCT116 cells. Sci. Rep. 2016, 6, 39123.
- Oh, E.T.; Kim, J.W.; Kim, J.M.; Kim, S.J.; Lee, J.S.; Hong, S.S.; Goodwin, J.; Ruthenborg, R.J.; Jung, M.G.; Lee, H.J.; et al. NQO1 inhibits proteasome-mediated degradation of HIF-1alpha. Nat. Commun. 2016, 7, 13593.
- Cummins, E.P.; Berra, E.; Comerford, K.M.; Ginouves, A.; Fitzgerald, K.T.; Seeballuck, F.; Godson, C.; Nielsen, J.E.; Moynagh, P.; Pouyssegur, J.; et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc. Natl. Acad. Sci. USA 2006, 103, 18154–18159.
- Fitzpatrick, S.F.; Tambuwala, M.M.; Bruning, U.; Schaible, B.; Scholz, C.C.; Byrne, A.; O’Connor, A.; Gallagher, W.M.; Lenihan, C.R.; Garvey, J.F.; et al. An intact canonical NF-kappaB pathway is required for inflammatory gene expression in response to hypoxia. J. Immunol. 2011, 186, 1091–1096.
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811.
- Walmsley, S.R.; Print, C.; Farahi, N.; Peyssonnaux, C.; Johnson, R.S.; Cramer, T.; Sobolewski, A.; Condliffe, A.M.; Cowburn, A.S.; Johnson, N.; et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J. Exp. Med. 2005, 201, 105–115.
- Ransone, L.J.; Verma, I.M. Nuclear proto-oncogenes Fos and Jun. Annu. Rev. Cell Biol. 1990, 6, 539–557.
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136.
- Nair, S.; Barve, A.; Khor, T.O.; Shen, G.X.; Lin, W.; Chan, J.Y.; Cai, L.; Kong, A.N. Regulation of Nrf2- and AP-1-mediated gene expression by epigallocatechin-3-gallate and sulforaphane in prostate of Nrf2-knockout or C57BL/6J mice and PC-3 AP-1 human prostate cancer cells. Acta Pharmacol. Sin. 2010, 31, 1223–1240.
- Fujioka, S.; Niu, J.; Schmidt, C.; Sclabas, G.M.; Peng, B.; Uwagawa, T.; Li, Z.; Evans, D.B.; Abbruzzese, J.L.; Chiao, P.J. NF-kappaB and AP-1 connection: Mechanism of NF-kappaB-dependent regulation of AP-1 activity. Mol. Cell. Biol. 2004, 24, 7806–7819.
- Verma, I.M.; Stevenson, J.K.; Schwarz, E.M.; Van Antwerp, D.; Miyamoto, S. Rel/NF-kappa B/I kappa B family: Intimate tales of association and dissociation. Genes Dev. 1995, 9, 2723–2735.
- Gong, H.; Tai, H.; Huang, N.; Xiao, P.; Mo, C.; Wang, X.; Han, X.; Zhou, J.; Chen, H.; Tang, X.; et al. Nrf2-SHP Cascade-Mediated STAT3 Inactivation Contributes to AMPK-Driven Protection Against Endotoxic Inflammation. Front. Immunol. 2020, 11, 414.
- Snyder, M.; Huang, J.; Huang, X.Y.; Zhang, J.J. A signal transducer and activator of transcription 3 Nuclear Factor kappaB (Stat3.NFkappaB) complex is necessary for the expression of fascin in metastatic breast cancer cells in response to interleukin (IL)-6 and tumor necrosis factor (TNF)-alpha. J. Biol. Chem. 2014, 289, 30082–30089.
- Arlt, A.; Schafer, H.; Kalthoff, H. The ‘N-factors’ in pancreatic cancer: Functional relevance of NF-kappaB, NFAT and Nrf2 in pancreatic cancer. Oncogenesis 2012, 1, e35.
- Serfling, E.; Berberich-Siebelt, F.; Avots, A.; Chuvpilo, S.; Klein-Hessling, S.; Jha, M.K.; Kondo, E.; Pagel, P.; Schulze-Luehrmann, J.; Palmetshofer, A. NFAT and NF-kappaB factors-the distant relatives. Int. J. Biochem. Cell Biol. 2004, 36, 1166–1170.
- Klotz, L.O.; Steinbrenner, H. Cellular adaptation to xenobiotics: Interplay between xenosensors, reactive oxygen species and FOXO transcription factors. Redox Biol. 2017, 13, 646–654.
- Lin, L.; Hron, J.D.; Peng, S.L. Regulation of NF-kappaB, Th activation, and autoinflammation by the forkhead transcription factor Foxo3a. Immunity 2004, 21, 203–213.


