1. Introduction
The panoply of mitochondrial functions reflects on highly heterogeneous clinical presentations when an error in a mitochondrial protein or function occurs. Mitochondria are dynamic and mobile organelles representing a hub where exchange of information among the nucleus and other cellular compartments takes place to modulate energy production and metabolites provision to the cell’s specific needs and nutrient availability
[1]. The basic function of mitochondria is the generation of more than 90% of cellular energy via the oxidative phosphorylation (OxPhos) system
[2] but, in addition, they play many roles in the different types of cells: compartmentalize metabolites for the maintenance of redox homeostasis; function as centers for metabolic waste management
[3]; surveil calcium homeostasis
[4]; initiate caspase-dependent apoptosis and other intermediate cellular stress response
[5]; provide sulfur metabolism and iron-sulfur cluster biogenesis
[6][7]; house the synthesis of cardiolipin, steroids, quinone, and heme
[8][9]; breakdown fatty acids through β-oxidation; and serve as a metabolic platform for the tricarboxylic acid (TCA), and urea cycles
[10]. All these functions include homeostatic regulation of organelle morphology and dynamics
[11], quality control
[12], and participation in the immune response
[13][14]. Alteration of each of the above functions and activities can have different effects according to the specificity of the organ and cell type, but alteration of mitochondrial energy production can impact tissues with the highest energy requirements such as the nervous system, both central (CNS) and peripheral (PNS)
[15][16].
The term ‘mitochondrial medicine’ categorizes the ample array of clinical presentations associated with all types of mitochondrial defects having directly or secondarily defect of one or several mitochondrial functions although ‘mitochondrial diseases’ traditionally indicate dysfunction of the OxPhos system
[6][17]. The direct link between human disease and the genetic alteration of a mitochondrial function has found a breakthrough with the application of -
omics technologies (i.e., genomics, transcriptomics, proteomics, metabolomics, and epigenomics, etc.). Rapidly, high-throughput
omics techniques—that is detection of biologically significant differences, even if not high magnitude changes, in a multitude of molecular constituents in organisms supported by sophisticated bioinformatics tools—have allowed progress in cataloging the predicted human mitochondrial proteins thus revealing new details and providing clues to elucidating still unknown basic aspects of mitochondrial structure and function. These novel high-throughput techniques have enhanced the final diagnosis of several mitochondrial disorders. This is a very relevant aspect, especially considering that mitochondrial diseases individually are rare but are probably the most frequent genetic disorder in adults (incidence of 1 in 5000 live births)
[18]. More recently, genome editing technology applied to neural cultures and cerebral organoids generated from patients-derived iPSCs is revolutionizing the landscape and offering new opportunities for understanding the pathogenetic effects of mutations in nervous tissue.
This review aims to focus on the dysfunction of OxPhos defects mostly in the nervous system to highlighting the contributions of powerful omics technologies to mitochondrial medicine to land from the laboratory to the clinic.
2. Mitoexome, Mitochondrial Proteome, and Mitointeractome
Before Next-Generation Sequencing (NGS) improved our understanding of how mutations cause diseases, first attempts to identify the mitochondrial proteome were based on ‘
cyberscreening’ of available genome databases. This allowed the discovery of few human mitochondrial genes presenting orthologs in lower eukaryotes. An example of the cyberscreening strategy used
Saccharomyces cerevisiae proteins as ‘probes’ to identify
BCS1,
PET112,
SCO1,
COX15, and
COX11, five yeast genes that present orthologs (respectively,
BCS1L,
GATB,
SCO1,
COX15, and
COX11) in humans
[19]. Except for
COX11, a COX-assembly, all genes have been implicated in mitochondrial diseases [OMIM 603647.0001-603647.0013; OMIM 603645.0001-603645.0002; OMIM 603644.0001-603644.0002; OMIM 603646.0001-603646.0004], see paragraphs 4.3 and 4.4. To date, whole-exome (WES) and whole-genome (WGS) resequencing have dramatically enhanced the ability to identify the underlying gene mutations in patients with isolated or multiple mitochondrial respiratory chain complex defects
[20][21]. The collection of mt genes and coding exons of the 1034 nuclear genes encoding the human mitochondrial proteome is defined as ‘MitoExome’
[22][23]. This multigene panel is useful in performing targeted resequencing of the OxPhos nuclear genes because it includes not only the 77 nuclear structural OxPhos subunits and the 37 mitochondrial (mt) DNA genes including the 13 structural genes for OxPhos subunits
[24] but also genes for mitochondrial proteins either already known or not to be associated with a specific mitochondrial disease, including assembly factors and electron carriers’ genes which represent a large fraction of the overall mitochondrial genes that can cause mitochondrial dysfunction
[21]. Application of
MitoExome resequencing provides novel mutation candidates, enables the discovery of unusual clinical variants
[25][26] and new clinical phenotypes
[26] (
Figure 1). Furthermore, the integration of MitoExome sequencing with the study of mitochondrial proteome potentiates the detection of variants causing protein destabilization and/or aberrantly low expression
[27].
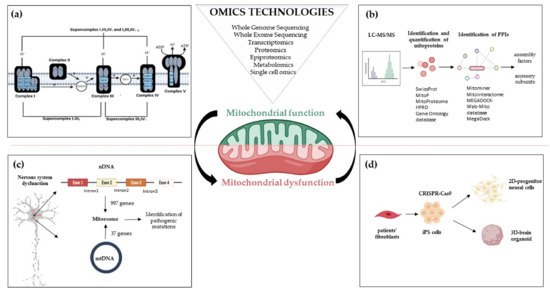
Figure 1. Omics strategies advance in understanding mitochondrial function and dysfunction in brain disorders related to OxPhos gene mutations. Mitochondrial bioenergetics involves activities whose function and structure have been deeply elucidated by omics technologies; (a) The introduction of high-resolution technologies has been resolutive to deepen the structure of the respiratory chain complexes and supercomplexes; (b) Quantitative proteomics, e.g., LC-MS/MS enable the identification and quantification of mitoproteins and provide large amounts of data. Through Network-based approaches analyzing protein-protein interactions, the huge amount of information allows the discovery of novel accessory subunits and assembly factors of the five multi-subunit enzyme complexes; (c) The re-sequencing carried out with MitoExome increases the possibility of identifying new or previously reported mutations in both mitochondrial and nuclear genes in patients; (d) Novel multi-omics analysis, based on single-cell omics, is applied to two-dimensional (2D) neural cultures and three-dimensional (3D) cerebral organoids generated from patients-derived iPSCs that can be engineered by CRISPR/Cas9. Abbreviations: LC-MS/MS: Liquid Chromatography with tandem mass spectrometry; PPIs: Protein-protein Interactions; nDNA: nuclear DNA; mtDNA: mitochondrial DNA; iPS cells: Induced Pluripotent Stem cells.
Biochemical and ultrastructural characterizations have uncovered the heterogeneity of mitochondria in their function, trafficking patterns, lifespan, and morphology across cell types and different cellular compartments. Different tissues, cell types, and cellular states have unique signatures of protein localization to mitochondria. In the proteomic comparison of the mitochondrial proteins, almost half are found as
core components in virtually all tissues, whereas the remaining are tissue-specific
[28][29]. The study of mitochondrial proteome starts with the isolation of mt compartment from cells and tissues and stands behind the availability of methodologies to isolate pure mitochondria from different sources to define exactly the function of each protein in each cell type of the human body
[30]. The performance of proteomics analysis is driven by the reduction of sample complexity, enhancement of mass spectrometry (MS) power of resolution, and the possibility to reduce the contamination of the sample with non-mitochondrial proteins owed to chemical and physical similarities between mitochondria and other cellular components (e.g., lysosomes). Since the initial rough estimates, it has been suggested that the mammalian mitochondrial proteome encompassed about 1000–1500 distinct proteins—including the 13 mtDNA-encoded proteins
[24]—that represent an important subset of the ~20,000 distinct mammalian proteins
[31][32] (
Figure 2).
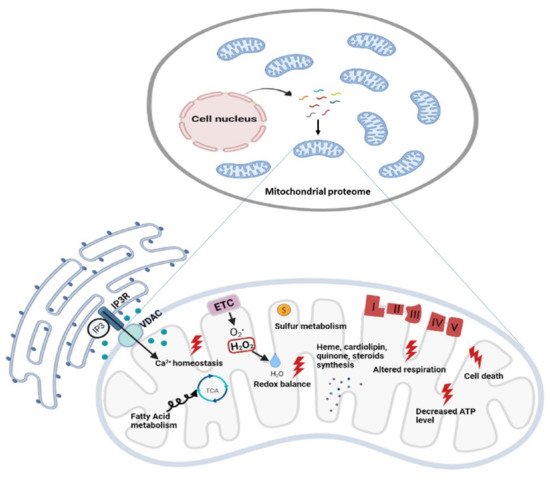
Figure 2. Functional diversity of mitochondrial proteins and bioenergetics consequences of OxPhos system dysfunction. The mammalian mitochondrial proteome includes both mitochondrial and nuclear DNA- encoded proteins. Most of the proteins required for the various activities in which mitochondria are involved are encoded by the nuclear genome, whereas the mitochondrial energy-producing system, i.e., the OxPhos complexes, has either mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) encoded components. The enlarged mitochondrion shows most of the bioenergetics consequences (indicated by the red bolt lightning) of genetic defects involving the OxPhos complexes. Abbreviations: IP3: Inositol Trisphosphate; IP3R: Inositol Trisphosphate Receptor; VDAC: Voltage-dependent anion channel; ETC: Electron Transport Chain; TCA: Tricarboxylic Acid Cycle; ATP: Adenosine triphosphate.
Quantitative two-dimensional (2D) gels of highly purified mitochondria estimated ~1500 distinct spots
[33], a number higher than the ~1000 distinct protein products encoded by the genomes of alpha-proteobacteria, which are the closest living relatives of modern-day mitochondria
[34]. Several databases have been used to integrate the experimental data with bioinformatic predictions based on mitochondrial localization or interaction. For example, the MitoProteome is an object-related database developed at the UCSD Supercomputer Center, which contains information on mitochondria-localized proteins
[35][36]. Each entry in the MitoProteome corresponds to a gene encoding a protein that is localized within mitochondria and its basic information, along with annotations of isoforms, splice variants, and functions of the corresponding protein. To date, the most comprehensive study elucidating the mitochondrial proteome of different mammalian tissues is represented by the MitoCarta inventory
[29][37]. This catalog combines multiple experimental and computational approaches, i.e., mass spectrometry (MS) analysis of mitochondria isolated from 14 mouse tissues, large-scale GFP-fusion microscopy analysis, and bioinformatics using data mining, prediction, evolutionary conservation, and a Bayesian integration of seven additional data sources. The first release was represented by MitoCarta1.0 (
http://www.broadinstitute.org/pubs/MitoCarta/; accessed 25 July 2021) which contained about 1000 distinct gene
loci [29]. Updated in 2016, MitoCarta 2.0 listed about 1200 genes
[37]. Another dedicated database that collected, curated, and annotated information on mitochondrial proteins is the
MitoMiner database (
http://mitominer.mrc-mbu.cam.ac.uk/; accessed 25 July 2021)
[38] (version 4.0, 2018). It is based on the literature and proteomics data based on both LC-MS and 2D gel studies, antibody staining, and other subcellular localization data, and provides a collective score for each protein’s probability to have the mitochondrial association. MitoMiner records mitochondrial proteins from 12 organisms
[38]. Using the data contained within MitoMiner, the
Integrated Mitochondrial Protein Index (IMPI) was also developed (
http://www.mrc-mbu.cam.ac.uk/impi; accessed 25 July 2021). IMPI version Q2 (2018) contains 1626 human genes that encode mitochondrially localized proteins, 1184 known to be mitochondrial and 442 predicted to be mitochondrial. The large amount of information provided by mito-databases as MitoMiner 4.0 v2018 JUN (
http://mitominer.mrc-mbu.cam.ac.uk; accessed 25 July 2021), makes it possible to define different score systems for mitochondrial confidence combining data from various mitochondrial and functional annotation databases. These strategies allow increasing the stringency of protein accepted as inherently mitochondrial
[39]. An exhaustive list of the major data sources loaded with the latest version and links to the relevant resources is reported in the Data Sources section of the Mitominer (
https://mitominer.mrc-mbu.cam.ac.uk/release-4.0/dataCategories.do; accessed 25 July 2021).
More recent advances in the experimental proteomic approaches, specifically in labeling and MS methods, have further expanded and defined the known mitochondrial proteome and have simultaneously revealed the sub-mitochondrial localization of many of them
[40][41]. A novel spatial proteomics pipeline demonstrated that many proteins cannot be classified to a single localization as they either transit between compartments or carry out their functional role(s) in multiple locations
[41]. The redundant functions, or functions affecting multiple cellular processes, rendered difficult the study and it was estimated that about ~20% of mitochondrial proteins remained uncharacterized
[42].
Along with technological progress that has enabled the discovery of approximately 78,120 human proteins [based on The UniProt Knowledgebase (UniProtKB), as of 23 February 2021], derives the challenge of identifying a large amount of potential protein-protein interactions (PPIs). An example of the network-based approaches analyzing protein-protein interaction is represented by MitoInteractome, a web-based portal containing 6549 protein sequences extracted from SwissProt (
http://www.expasy.ch/sprot/; accessed 25 July 2021), MitoP (
http://www.mitop.de:8080/mitop2/; accessed 25 July 2021), MitoProteome (
http://www.mitoproteome.org/; accessed 25 July 2021), HPRD (
http://www.hprd.org; accessed 25 July 2021) and Gene Ontology database (
http://www.geneontology.org; accessed 25 July 2021). This enables the elucidation of integrative mitochondrial functions and can expedite the discovery of novel interactions which otherwise may have been missed using traditional experimental techniques. MEGADOCK
[43][44], a structure-based PPI prediction method, was first developed and then followed the MEGADOCK-Web-Mito database which is a PPI prediction data archive, that includes prediction results for protein pairs of 654 mitochondria-related human proteins
[45]. All these approaches have been key in the study of PPI as a means to infer functions for uncharacterized proteins and to enable the discovery of novel proteins, e.g., several complex I assembly factors
[46][47] (
Figure 1).
For expert reviews on the details about the technical approaches, the required bioinformatics pipelines, and how (multi)omics technologies can help in studying the dysfunction of mitochondrial bioenergetics, see
[48][49].
 +1 point
+1 point