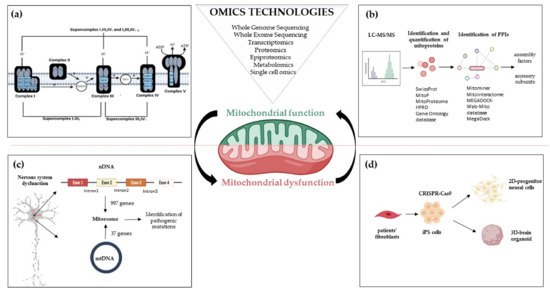
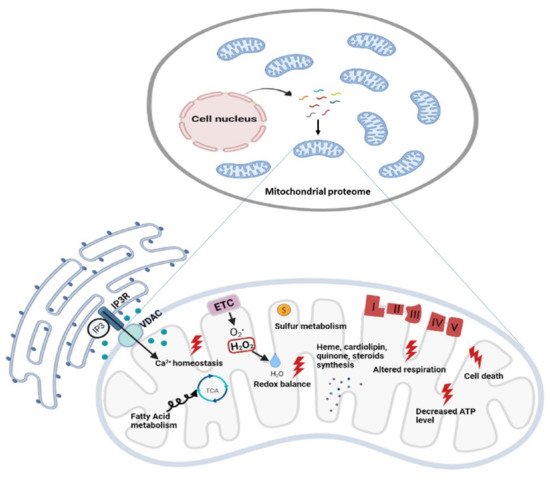
Oxidative phosphorylation (OxPhos) is the basic function of mitochondria, although the landscape of mitochondrial functions is continuously growing to include more aspects of cellular homeostasis. Thanks to the application of -omics technologies to the study of the OxPhos system, novel features emerge from the cataloging of novel proteins as mitochondrial thus adding details to the mitochondrial proteome and defining novel metabolic cellular interrelations, especially in the human brain. We focussed on the diversity of bioenergetics demand and different aspects of mitochondrial structure, functions, and dysfunction in the brain. Definition such as ‘mitoexome’, ‘mitoproteome’ and ‘mitointeractome’ have entered the field of ‘mitochondrial medicine’.
- mitochondria
- mitochondrial DNA
- nervous tissue
1. Introduction
2. Mitoexome, Mitochondrial Proteome, and Mitointeractome
References
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34.
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–148.
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754.
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340.
- Green, D.R.; Galluzzi, L.; Kroemer, G. Metabolic control of cell death. Science 2014, 345, 1250256.
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080.
- Muraresku, C.C.; McCormick, E.M.; Falk, M.J. Mitochondrial disease: Advances in Clinical diagnosis, management, therapeutic development, and preventative strategies. Curr. Genet. Med. Rep. 2018, 6, 62–72.
- Piel, R.B.; Dailey, H.A.; Medlock, A.E. The mitochondrial heme metabolon: Insights into the complex(Ity) of heme synthesis and distribution. Mol. Genet. Metab. 2019, 128, 198–203.
- Miller, W.L. Steroid hormone synthesis in mitochondria. Mol. Cell. Endocrinol. 2013, 379, 62–73.
- Anderson, A.J.; Jackson, T.D.; Stroud, D.A.; Stojanovski, D. Mitochondria—Hubs for regulating cellular biochemistry: Emerging concepts and networks. Open Biol. 2019, 9, 190126.
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287.
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185.
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498.
- Tiku, V.; Tan, M.-W.; Dikic, I. Mitochondrial functions in infection and immunity. Trends Cell Biol. 2020, 30, 263–275.
- Schapira, A.H. Mitochondrial disease. Lancet 2006, 368, 70–82.
- DiMauro, S.; Schon, E.A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 2003, 348, 2656–2668.
- Luft, R. The development of mitochondrial medicine. Proc. Natl. Acad. Sci. USA 1994, 91, 8731–8738.
- La Morgia, C.; Maresca, A.; Caporali, L.; Valentino, M.L.; Carelli, V. Mitochondrial diseases in adults. J. Intern. Med. 2020, 287, 592–608.
- Petruzzella, V.; Tiranti, V.; Fernandez, P.; Ianna, P.; Carrozzo, R.; Zeviani, M. Identification and characterization of human CDNAs specific to BCS1, PET112, SCO1, COX15, and COX11—Five genes involved in the formation and function of the mitochondrial respiratory chain. Genomics 1998, 54, 494–504.
- Stenton, S.L.; Prokisch, H. Advancing genomic approaches to the molecular diagnosis of mitochondrial disease. Essays Biochem. 2018, 62, 399–408.
- Stenton, S.L.; Prokisch, H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine 2020, 56, 102784.
- Calvo, S.E.; Compton, A.G.; Hershman, S.G.; Lim, S.C.; Lieber, D.S.; Tucker, E.J.; Laskowski, A.; Garone, C.; Liu, S.; Jaffe, D.B.; et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med. 2012, 4, 118ra10.
- Plutino, M.; Chaussenot, A.; Rouzier, C.; Ait-El-Mkadem, S.; Fragaki, K.; Paquis-Flucklinger, V.; Bannwarth, S. Targeted next generation sequencing with an extended gene panel does not impact variant detection in mitochondrial diseases. BMC Med. Genet. 2018, 19, 57.
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465.
- Garone, C.; Donati, M.A.; Sacchini, M.; Garcia-Diaz, B.; Bruno, C.; Calvo, S.; Mootha, V.K.; DiMauro, S. Mitochondrial encephalomyopathy due to a novel mutation in ACAD9. JAMA Neurol. 2013, 70, 1177–1179.
- Oláhová, M.; Berti, C.C.; Collier, J.J.; Alston, C.L.; Jameson, E.; Jones, S.A.; Edwards, N.; He, L.; Chinnery, P.F.; Horvath, R.; et al. Molecular genetic investigations identify new clinical phenotypes associated with BCS1L-related mitochondrial disease. Hum. Mol. Genet. 2019, 28, 3766–3776.
- Stenton, S.L.; Kremer, L.S.; Kopajtich, R.; Ludwig, C.; Prokisch, H. The diagnosis of inborn errors of metabolism by an integrative “multi-omics’’ approach: A perspective encompassing genomics, transcriptomics, and proteomics. J. Inherit. Metab. Dis. 2020, 43, 25–35.
- Mootha, V.K.; Bunkenborg, J.; Olsen, J.V.; Hjerrild, M.; Wisniewski, J.R.; Stahl, E.; Bolouri, M.S.; Ray, H.N.; Sihag, S.; Kamal, M.; et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 2003, 115, 629–640.
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.-E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123.
- Gonczarowska-Jorge, H.; Zahedi, R.P.; Sickmann, A. The proteome of baker’s yeast mitochondria. Mitochondrion 2017, 33, 15–21.
- Clamp, M.; Fry, B.; Kamal, M.; Xie, X.; Cuff, J.; Lin, M.F.; Kellis, M.; Lindblad-Toh, K.; Lander, E.S. Distinguishing protein-coding and noncoding genes in the human genome. Proc. Natl. Acad. Sci. USA 2007, 104, 19428–19433.
- Ponomarenko, E.A.; Poverennaya, E.V.; Ilgisonis, E.V.; Pyatnitskiy, M.A.; Kopylov, A.T.; Zgoda, V.G.; Lisitsa, A.V.; Archakov, A.I. The size of the human proteome: The width and depth. Int. J. Anal. Chem. 2016, 2016, 7436849.
- Lopez, M.F.; Kristal, B.S.; Chernokalskaya, E.; Lazarev, A.; Shestopalov, A.I.; Bogdanova, A.; Robinson, M. High-throughput profiling of the mitochondrial proteome using affinity fractionation and automation. Electrophoresis 2000, 21, 3427–3440.
- Karlberg, O.; Canbäck, B.; Kurland, C.G.; Andersson, S.G. The dual origin of the yeast mitochondrial proteome. Yeast 2000, 17, 170–187.
- Cotter, D. MitoProteome: Mitochondrial protein sequence database and annotation system. Nucleic Acids Res. 2004, 32, D463–D467.
- Guda, P.; Subramaniam, S.; Guda, C. Mitoproteome: Human heart mitochondrial protein sequence database. Methods Mol Biol. 2007, 357, 375–383.
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257.
- Smith, A.C.; Robinson, A.J. MitoMiner v3.1, an update on the mitochondrial proteomics database. Nucleic Acids Res. 2016, 44, D1258–D1261.
- Doccini, S.; Morani, F.; Nesti, C.; Pezzini, F.; Calza, G.; Soliymani, R.; Signore, G.; Rocchiccioli, S.; Kanninen, K.M.; Huuskonen, M.T.; et al. Proteomic and functional analyses in disease models reveal CLN5 protein involvement in mitochondrial dysfunction. Cell Death Discov. 2020, 6, 18.
- Hung, V.; Lam, S.S.; Udeshi, N.D.; Svinkina, T.; Guzman, G.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Correction: Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. eLife 2019, 8, e50707.
- Geladaki, A.; Kočevar Britovšek, N.; Breckels, L.M.; Smith, T.S.; Vennard, O.L.; Mulvey, C.M.; Crook, O.M.; Gatto, L.; Lilley, K.S. Combining LOPIT with differential ultracentrifugation for high-resolution spatial proteomics. Nat. Commun. 2019, 10, 331.
- Sung, A.Y.; Floyd, B.J.; Pagliarini, D.J. Systems biochemistry approaches to defining mitochondrial protein function. Cell Metab. 2020, 31, 669–678.
- Ohue, M.; Matsuzaki, Y.; Uchikoga, N.; Ishida, T.; Akiyama, Y. MEGADOCK: An all-to-all protein-protein interaction prediction system using tertiary structure data. Protein Pept. Lett. 2013, 21, 766–778.
- Ohue, M.; Shimoda, T.; Suzuki, S.; Matsuzaki, Y.; Ishida, T.; Akiyama, Y. MEGADOCK 4.0: An ultra–high-performance protein–protein docking software for heterogeneous supercomputers. Bioinformatics 2014, 30, 3281–3283.
- Hayashi, T.; Matsuzaki, Y.; Yanagisawa, K.; Ohue, M.; Akiyama, Y. MEGADOCK-Web: An integrated database of high-throughput structure-based protein-protein interaction predictions. BMC Bioinform. 2018, 19, 62.
- Floyd, B.J.; Wilkerson, E.M.; Veling, M.T.; Minogue, C.E.; Xia, C.; Beebe, E.T.; Wrobel, R.L.; Cho, H.; Kremer, L.S.; Alston, C.L.; et al. Mitochondrial protein interaction mapping identifies regulators of respiratory chain function. Mol. Cell 2016, 63, 621–632.
- Formosa, L.E.; Dibley, M.G.; Stroud, D.A.; Ryan, M.T. Building a complex complex: Assembly of mitochondrial respiratory chain complex I. Semin. Cell Dev. Biol. 2018, 76, 154–162.
- Labory, J.; Fierville, M.; Ait-El-Mkadem, S.; Bannwarth, S.; Paquis-Flucklinger, V.; Bottini, S. Multi-omics approaches to improve mitochondrial disease diagnosis: Challenges, advances, and perspectives. Front. Mol. Biosci. 2020, 7, 590842.
- Khan, S.; Ince-Dunn, G.; Suomalainen, A.; Elo, L.L. Integrative omics approaches provide biological and clinical insights: Examples from mitochondrial diseases. J. Clin. Investig. 2020, 130, 20–28.