 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Shannon Erhardt | + 1817 word(s) | 1817 | 2021-08-03 12:17:34 |
Video Upload Options
Cardiac neural crest cells (NCCs), a specified subpopulation of the neural crest (NC), are vital for normal cardiovascular development, as they significantly contribute to the pharyngeal arch arteries, the developing cardiac outflow tract (OFT), cardiac valves, and interventricular septum. Various signaling pathways and factors are shown to orchestrate the proper migration, compaction, and differentiation of cardiac NCCs during cardiovascular development. Any loss or dysregulation of various signaling components in cardiac NCCs can lead to abnormal cardiovascular development during embryogenesis, resulting in abnormalities categorized as congenital heart defects (CHDs).
1. Introduction
Neural crest cells (NCCs) are a multipotent, and highly migratory, transient vertebrate cell population originating in the dorsal region of the neural tube. During embryogenesis, NCCs, upon neural plate folding, arise from either side of the neural plate at a region called the neural plate border, situated between the neuroectoderm and non-neuroectoderm [1][2]. During and after the neural plate closes, NCCs undergo epithelial-to-mesenchymal transition (EMT) in which they obtain their migratory potential and disperse from the neural tube, relocating to specific locations throughout the embryo, to differentiate into a wide variety of cell types, such as osteoblasts and smooth muscle cells [2][3][4]. Although NCCs arise sequentially during embryo development, they are specified into four main subpopulations based on their anteroposterior axis position, differential abilities, and corresponding terminal locations [5]: cranial neural crest (NC), contributing to the majority of bone and cartilage formation of the head [6]; vagal NC, aiding in the formation of the thymus, lung, enteric nervous system and cardiovascular system [7]; trunk NC, contributing to the peripheral nervous and endocrine systems [8]; sacral NC, aiding in the development of neurons and glia of the enteric nervous system [9]. However, studies on these subpopulations have indicated that the vagal NC consists of a smaller specified group of cells deemed the cardiac NC, known to significantly contribute to cardiovascular development, along with aiding in the development of the thymus, thyroid glands, and cardiac ganglia [7][10][11][12][13].
NC ablation studies have demonstrated the importance of NCCs for proper heart formation. Deficiencies in these cells result in a variety of cardiac malformations during embryonic development categorized as congenital heart defects (CHDs) [14]. CHDs are the most common birth defect and the leading cause of birth defect-related deaths [15][16][17], with a variety of phenotypes ranging from mild forms, accompanied by minimal cardiac complications, to severe and life-threatening forms, resulting in extreme cardiac impediments and death. The most common defects, seen in approximately 30% of human cases, are atrial and ventricular septal defects (ASD/VSD), caused by a hole along the interatrial septum or interventricular septum, respectively [18]. Other common CHDs include malformations of the cardiac outflow tract (OFT), which normally gives rise to the proper vessel development and septation of the semilunar valves (leading to the aorta and pulmonary valves). OFT defects result in common CHD phenotypes such as transposition of the great arteries (TGA), aortic or pulmonary artery stenosis, and patent ductus arteriosus (PDA), as well as rarer CHD phenotypes, such as persistent truncus arteriosus (PTA), characterized by the absence of aortopulmonary septation, overriding aorta, in which the aorta is centralized over the VSD and open to both ventricles, and double outlet right ventricle (DORV), where the aorta is connected to the right, instead of left, ventricle [19][20][21]. Multiple congenital human syndromes, such as CHARGE, Treacher Collins, and DiGeorge, are associated with a variety of CHD phenotypes. Although substantial progress has been made in uncovering the origin of congenital cardiac diseases, details regarding how NCCs contribute to heart development, and how NC deficiencies cause CHDs, is still under investigation.
2. Neural Crest Associated Cardiac Congenital Abnormalities in Humans
Cardiac NC deficiency phenotypes are consistent with the CHDs present in a variety of human diseases. In recent years, the prevalence of CHDs has increased due to improved pediatric screening and increased survival [18]. However, further investigation is needed to determine the pathogenic and/or novel genetic deficiencies contributing to these cardiac defects, to improve diagnostic screening and treatment. Although many CHDs are considered nonsyndromic, multiple genetic syndromes, including DiGeorge and CHARGE, involve NC deficiencies with known associated cardiac phenotypes. Not only do NC deficiencies produce heart defects, they are also known to contribute to various craniofacial defects common among human cardio-craniofacial syndromes, such as the most common multiple anomaly syndromes DiGeorge, Noonan, and Velo-cardio-facial [22][23]. However, more studies are needed to determine how cardiac NC contributions are similar and/or different between multiple syndromes, and what NC regulatory pathways and factors are altered in such diseases.
2.1. DiGeorge Syndrome
DiGeorge syndrome, also known as 22q11.2 deletion, has a well-defined phenotype consisting of characteristic facial features, immunodeficiencies, CHDs, hypocalcemia, and developmental delays. Commonly associated CHDs include interrupted aortic arch, VSD, TOF, and PTA [24][25][26]. Tbx1, a region of Df1, the first targeted region homologous to human 22q11, is expressed in the pharyngeal region and is necessary for OFT and aortic arch development [27][28]. Although Tbx1 has shown not to be expressed by NCCs [27][29], the loss of Tbx1 has shown to be associated with VSD and tetralogy of fallot (TOF) in patients [30]. Calmont and colleagues found that in mice, Tbx1 drives downstream expression of genes such as Gbx2, to regulate cardiac NCC migration to the pharyngeal arches by a non-cell-autonomous effect, and that the combinational disruption of Tbx1 and Gbx2 results in abnormal pharyngeal arch development, along with aortic arch interruption (Figure 1), suggestively due to cardiac NC migration deficiencies [31]. However, further studies are needed to determine the specifics regarding the onset and longevity of such NC migration deficiencies, along with determining whether other various CHD phenotypes arise. It has been shown that a loss of Tbx1 in mice, negatively impacts the development of the second heart field (SHF), partly due to a lack of cell proliferation, which can produce OFT hypoplasia and vascular trunk mislocalization [32][33]. SHF progenitors are a multipotent cardiac progenitor population important for heart tube looping, along with myocardium, smooth muscle, and endothelial cell formation. Given that SHF progenitors and cardiac NCCs closely interact and make major contributions to the development of the OFT, it is of great interest to determine the impact of such Tbx1 depletions in the SHF on cardiac NCCs during heart development.
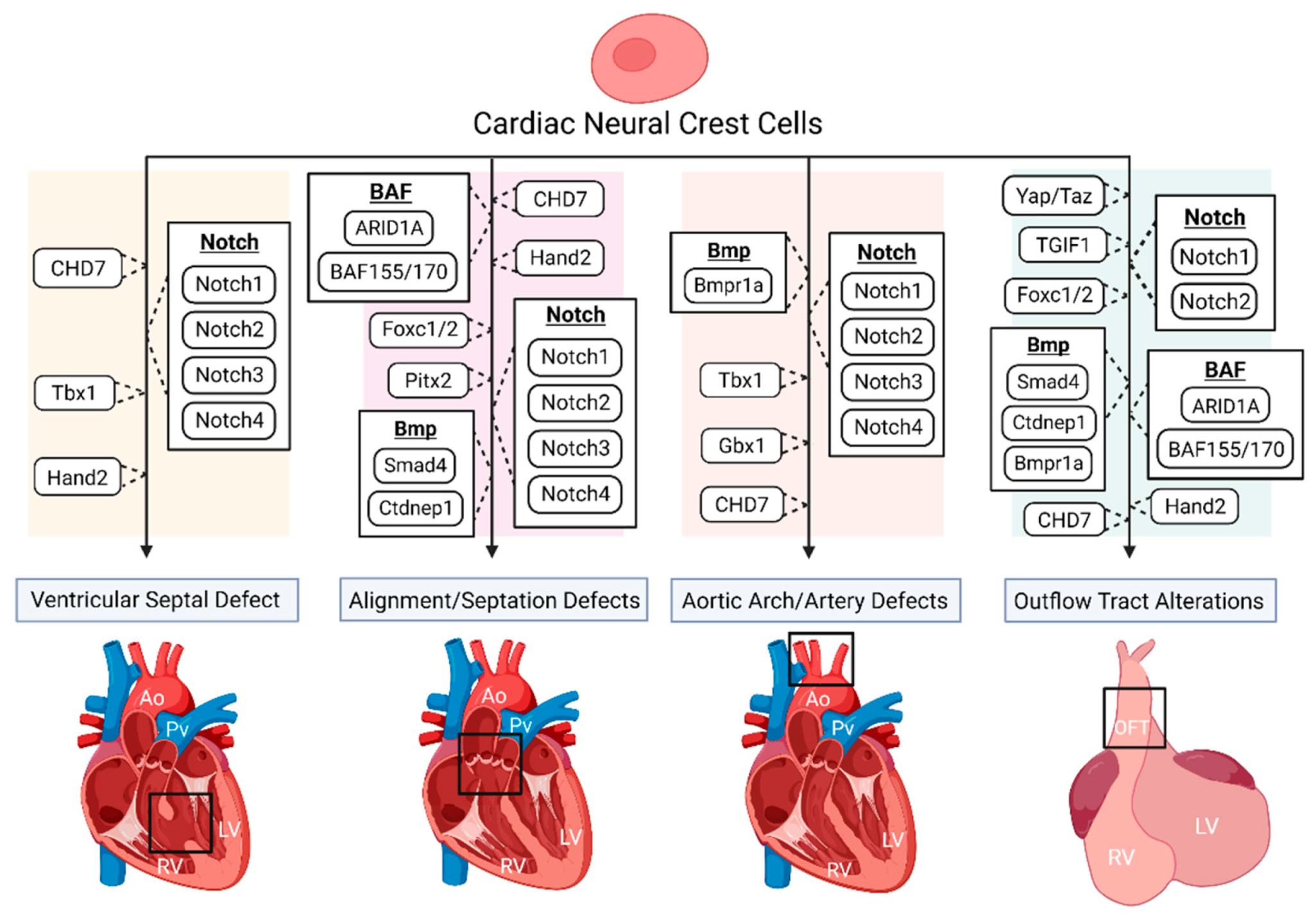 Figure 1. The disruption of various genes and signaling pathways, indicated to be important for proper cardiac neural crest (NC) contribution, results in numerous congenital heart defect (CHD) phenotypes.
Figure 1. The disruption of various genes and signaling pathways, indicated to be important for proper cardiac neural crest (NC) contribution, results in numerous congenital heart defect (CHD) phenotypes.
2.2. CHARGE Syndrome
CHARGE syndrome affects multiple organ systems and is an acronym for coloboma, heart defects, atresia choanae, growth and mental retardation, genital abnormalities, and ear abnormalities [34]. The most common CHD associated with CHARGE is TOF, detected in approximately 33% of human cases, followed by VSD and aortic arch abnormalities, suggesting that NC development is possibly affected during embryogenesis [35]. A study by Bajpai and colleagues showed that CHD7, the only mutated gene known to cause CHARGE syndrome, is essential for NC migration and the promotion of key transcriptional regulatory genes such as Sox9, Twist1, and Slug, both in vivo (Xenopus) and in vitro (human embryonic stem cells), while a deficiency of CHD7 caused cardiac OFT defects in Xenopus embryos, resulting in vascular septation defects such as PTA (Figure 1) [36]. A recent study by Yan and colleagues found that the deletion of CHD7 in NCCs of mice, by the use of the Wnt1-Cre2 neural crest-specific driver, not only results in severe conotruncal defects (VSD and DORV), interrupted aortic arch, and perinatal lethality, but inhibits the OFT invasion of cardiac NCCs (Figure 1) [37]. The authors suggest that such cardiac defects are due to the establishment that CHD7 directly regulates key NC regulatory genes such as Foxc2 and Hand2, through ATP-dependent and -independent functions, clarifying the molecular etiology of CHD7-related cardiac defects [37].
2.3. Treacher Collins Syndrome
Cardiac malformations can occur in patients with Treacher Collins syndrome. Treacher Collins is most commonly caused by a mutation within the TCOF1 gene. TCOF1 is involved in mRNA formation in NCCs during embryogenesis, largely associated with NC depletions in pharyngeal arches 1 and 2 [38]. Treacher Collins is normally associated with craniofacial abnormalities, but the depletion of NCCs has also been indicated in producing human CHD phenotypes such as VSD, ASD, and PDA [39]. Studies have found that haploinsufficiency of TCOF1 in mice results in a reduced number of migrating NCCs, leading to severe craniofacial defects [38]. More recently, Sanchez and colleagues found that the knockdown of one of the only genes known to be involved in Treacher Collins, POLR1B (RNA polymerase1 subunit B), results in notable cardiac edema, reduced NC migration, and embryonic death in zebrafish [40]. Serrano and colleagues, using human pluripotent stem cell (HPSC)-derived NCCs, found that disruption of TCOF1, by siRNA, confirmed previous conclusions that NC migration is impaired, but also found that NC proliferation was reduced [41]. Although many in vivo TCOF1 studies focus on cranial NC contributions, their findings, along with those from in vitro experiments, indicate a potential role of TCOF1 in cardiac NC function and cardiac formation, which requires further investigation.
3. Conclusions and Future Perspective
References
- Xi, M.; Lui, F. Neuroanatomy, Neural Crest. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021.
- Sauka-Spengler, T.; Bronner-Fraser, M. A gene regulatory network orchestrates neural crest formation. Nat. Rev. Mol. Cell Biol. 2008, 9, 557–568.
- Arima, Y.; Miyagawa-Tomita, S.; Maeda, K.; Asai, R.; Seya, D.; Minoux, M.; Rijli, F.M.; Nishiyama, K.; Kim, K.S.; Uchijima, Y.; et al. Preotic neural crest cells contribute to coronary artery smooth muscle involving endothelin signalling. Nat. Commun. 2012, 3, 1267.
- Etchevers, H.C.; Dupin, E.; Le Douarin, N.M. The diverse neural crest: From embryology to human pathology. Development 2019, 146.
- Rocha, M.; Beiriger, A.; Kushkowski, E.E.; Miyashita, T.; Singh, N.; Venkataraman, V.; Prince, V.E. From head to tail: Regionalization of the neural crest. Development 2020, 147.
- Wu, T.; Chen, G.; Tian, F.; Liu, H.X. Contribution of cranial neural crest cells to mouse skull development. Int. J. Dev. Biol. 2017, 61, 495–503.
- Kirby, M.L.; Gale, T.F.; Stewart, D.E. Neural crest cells contribute to normal aorticopulmonary septation. Science 1983, 220, 1059–1061.
- Weston, J.A. A radioautographic analysis of the migration and localization of trunk neural crest cells in the chick. Dev. Biol. 1963, 6, 279–310.
- Pomeranz, H.D.; Rothman, T.P.; Gershon, M.D. Colonization of the post-umbilical bowel by cells derived from the sacral neural crest: Direct tracing of cell migration using an intercalating probe and a replication-deficient retrovirus. Development 1991, 111, 647–655.
- Kirby, M.L. Cardiac morphogenesis—Recent research advances. Pediatric Res. 1987, 21, 219–224.
- Verberne, M.E.; Gittenberger-de Groot, A.C.; van Iperen, L.; Poelmann, R.E. Distribution of different regions of cardiac neural crest in the extrinsic and the intrinsic cardiac nervous system. Dev. Dyn. 2000, 217, 191–204.
- Kirby, M.L.; Stewart, D.E. Neural crest origin of cardiac ganglion cells in the chick embryo: Identification and extirpation. Dev. Biol. 1983, 97, 433–443.
- Hutchins, E.J.; Kunttas, E.; Piacentino, M.L.; Howard, A.G.A.t.; Bronner, M.E.; Uribe, R.A. Migration and diversification of the vagal neural crest. Dev. Biol. 2018, 444 (Suppl. 1), S98–S109.
- Kirby, M.L. Cardiac Development; Oxford University Press: Oxford, UK; New York, NY, USA, 2007.
- Van der Linde, D.; Konings, E.E.; Slager, M.A.; Witsenburg, M.; Helbing, W.A.; Takkenberg, J.J.; Roos-Hesselink, J.W. Birth prevalence of congenital heart disease worldwide: A systematic review and meta-analysis. J. Am. Coll. Cardiol. 2011, 58, 2241–2247.
- Bouma, B.J.; Mulder, B.J. Changing Landscape of Congenital Heart Disease. Circ. Res. 2017, 120, 908–922.
- Liu, Y.; Chen, S.; Zuhlke, L.; Black, G.C.; Choy, M.K.; Li, N.; Keavney, B.D. Global birth prevalence of congenital heart defects 1970–2017: Updated systematic review and meta-analysis of 260 studies. Int. J. Epidemiol. 2019, 48, 455–463.
- Wu, W.; He, J.; Shao, X. Incidence and mortality trend of congenital heart disease at the global, regional, and national level, 1990–2017. Medicine 2020, 99, e20593.
- Williams, J.M.; de Leeuw, M.; Black, M.D.; Freedom, R.M.; Williams, W.G.; McCrindle, B.W. Factors associated with outcomes of persistent truncus arteriosus. J. Am. Coll. Cardiol. 1999, 34, 545–553.
- Pradat, P.; Francannet, C.; Harris, J.A.; Robert, E. The epidemiology of cardiovascular defects, part I: A study based on data from three large registries of congenital malformations. Pediatric Cardiol. 2003, 24, 195–221.
- Yin, W.-h.; Lu, B. Tetralogy of Fallot (TOF). In Cardiac CT: Diagnostic Guide and Cases; Jin, Z.-Y., Lu, B., Wang, Y., Eds.; Springer: Singapore, 2020; pp. 103–108.
- Hutson, M.R.; Kirby, M.L. Model systems for the study of heart development and disease. Cardiac neural crest and conotruncal malformations. Semin Cell Dev. Biol. 2007, 18, 101–110.
- Tartaglia, M.; Gelb, B.D.; Zenker, M. Noonan syndrome and clinically related disorders. Best Pr. Res. Clin. Endocrinol. Metab. 2011, 25, 161–179.
- Carotti, A.; Digilio, M.C.; Piacentini, G.; Saffirio, C.; Di Donato, R.M.; Marino, B. Cardiac defects and results of cardiac surgery in 22q11.2 deletion syndrome. Dev. Disabil. Res. Rev. 2008, 14, 35–42.
- Shprintzen, R.J. Velo-cardio-facial syndrome: 30 Years of study. Dev. Disabil. Res. Rev. 2008, 14, 3–10.
- Momma, K. Cardiovascular anomalies associated with chromosome 22q11.2 deletion syndrome. Am. J. Cardiol. 2010, 105, 1617–1624.
- Lindsay, E.A.; Vitelli, F.; Su, H.; Morishima, M.; Huynh, T.; Pramparo, T.; Jurecic, V.; Ogunrinu, G.; Sutherland, H.F.; Scambler, P.J.; et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 2001, 410, 97–101.
- Papangeli, I.; Scambler, P. The 22q11 deletion: DiGeorge and velocardiofacial syndromes and the role of TBX1. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 393–403.
- Vitelli, F.; Morishima, M.; Taddei, I.; Lindsay, E.A.; Baldini, A. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Hum. Mol. Genet. 2002, 11, 915–922.
- Ganji, H.; Salehi, M.; Sedghi, M.; Abdali, H.; Nouri, N.; Sadri, L.; Hosseinzadeh, M.; Vakili, B.; Lotfi, M. Investigation of TBX1 gene deletion in Iranian children with 22q11.2 deletion syndrome: Correlation with conotruncal heart defects. Heart Asia 2013, 5, 200–202.
- Calmont, A.; Ivins, S.; Van Bueren, K.L.; Papangeli, I.; Kyriakopoulou, V.; Andrews, W.D.; Martin, J.F.; Moon, A.M.; Illingworth, E.A.; Basson, M.A.; et al. Tbx1 controls cardiac neural crest cell migration during arch artery development by regulating Gbx2 expression in the pharyngeal ectoderm. Development 2009, 136, 3173–3183.
- Theveniau-Ruissy, M.; Dandonneau, M.; Mesbah, K.; Ghez, O.; Mattei, M.G.; Miquerol, L.; Kelly, R.G. The del22q11.2 candidate gene Tbx1 controls regional outflow tract identity and coronary artery patterning. Circ. Res. 2008, 103, 142–148.
- Kelly, R.G.; Papaioannou, V.E. Visualization of outflow tract development in the absence of Tbx1 using an FgF10 enhancer trap transgene. Dev. Dyn. 2007, 236, 821–828.
- Yasuda, K.; Morihana, E.; Fusazaki, N.; Ishikawa, S. Cardiovascular Malformations in CHARGE Syndrome with DiGeorge Phenotype: Two Case Reports. Case Rep. Pediatrics 2016, 2016, 8013530.
- Corsten-Janssen, N.; Kerstjens-Frederikse, W.S.; du Marchie Sarvaas, G.J.; Baardman, M.E.; Bakker, M.K.; Bergman, J.E.; Hove, H.D.; Heimdal, K.R.; Rustad, C.F.; Hennekam, R.C.; et al. The cardiac phenotype in patients with a CHD7 mutation. Circ. Cardiovasc. Genet. 2013, 6, 248–254.
- Bajpai, R.; Chen, D.A.; Rada-Iglesias, A.; Zhang, J.; Xiong, Y.; Helms, J.; Chang, C.P.; Zhao, Y.; Swigut, T.; Wysocka, J. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 2010, 463, 958–962.
- Yan, S.; Thienthanasit, R.; Chen, D.; Engelen, E.; Bruhl, J.; Crossman, D.K.; Kesterson, R.; Wang, Q.; Bouazoune, K.; Jiao, K. CHD7 regulates cardiovascular development through ATP-dependent and -independent activities. Proc. Natl. Acad. Sci. USA 2020, 117, 28847–28858.
- Dixon, J.; Jones, N.C.; Sandell, L.L.; Jayasinghe, S.M.; Crane, J.; Rey, J.P.; Dixon, M.J.; Trainor, P.A. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc. Natl. Acad. Sci. USA 2006, 103, 13403–13408.
- Dobrilovic, N.; Fernandez, A.B.; Lin, A.; Singh, A.K. Treacher Collins syndrome: Sinus of Valsalva aneurysm. Circulation 2013, 128, e12–e13.
- Sanchez, E.; Laplace-Builhe, B.; Mau-Them, F.T.; Richard, E.; Goldenberg, A.; Toler, T.L.; Guignard, T.; Gatinois, V.; Vincent, M.; Blanchet, C.; et al. POLR1B and neural crest cell anomalies in Treacher Collins syndrome type 4. Genet. Med. Off. J. Am. Coll. Med. Genet. 2020, 22, 547–556.
- Serrano, F.; Bernard, W.G.; Granata, A.; Iyer, D.; Steventon, B.; Kim, M.; Vallier, L.; Gambardella, L.; Sinha, S. A Novel Human Pluripotent Stem Cell-Derived Neural Crest Model of Treacher Collins Syndrome Shows Defects in Cell Death and Migration. Stem. Cells Dev. 2019, 28, 81–100.
- Lev, M. Conduction system in congenital heart disease. Am. J. Cardiol. 1968, 21, 619–627.
- Cavanaugh, A.M.; Huang, J.; Chen, J.N. Two developmentally distinct populations of neural crest cells contribute to the zebrafish heart. Dev. Biol. 2015, 404, 103–112.
- Sato, M.; Yost, H.J. Cardiac neural crest contributes to cardiomyogenesis in zebrafish. Dev. Biol. 2003, 257, 127–139.


