In recent decades, fluorescent microscopy has become the most popular tool of cell and developmental biologists, since fluorescent labeling offers high molecular specificity, and due to its low invasiveness, it has become the leading method to study living cells, tissues or organisms. In the early days, only widefield applications were available, but with later technological advances, every aspect of microscopy was improved tremendously, resulting in the development of specialized microscopes, such as optical sectioning confocal microscopes, spinning disk confocal microscopes, light sheets, TIRF and multi-photon microscopic tools. Despite the remarkable progress, all these techniques remained limited by the wavelength of light at around 200-250 nm. A turning point came when scientists found ways to get around this obstacle and developed several nanoscopic methods that are able to increase the resolution up to 2-10 nm, allowing the study and understanding of the cellular structures at the molecular level by broadly applicable microscopic approaches.
1. Overview
With the advent of super-resolution microscopy, we gained a powerful toolbox to bridge the gap between the cellular- and molecular-level analysis of living organisms. Although nanoscopy is broadly applicable, classical model organisms, such as fruit flies, worms and mice, remained the leading subjects because combining the strength of sophisticated genetics, biochemistry and electrophysiology with the unparalleled resolution provided by super-resolution imaging appears as one of the most efficient approaches to understanding the basic cell biological questions and the molecular complexity of life. Here, we summarize the major nanoscopic techniques and illustrate how these approaches were used in Drosophila model systems to revisit a series of well-known cell biological phenomena. These investigations clearly demonstrate that instead of simply achieving an improvement in image quality, nanoscopy goes far beyond with its immense potential to discover novel structural and mechanistic aspects. With the examples of synaptic active zones, centrosomes and sarcomeres, we will explain the instrumental role of super-resolution imaging pioneered in Drosophila in understanding fundamental subcellular constituents.
2. Breaking the diffraction limit
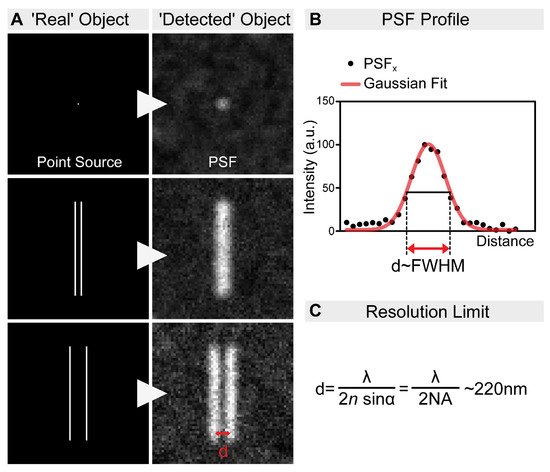
Today’s scientists wish to understand the biological processes at a molecular level. Fluorescent microscopy, allowing the labeling of biomolecules with high specificity by the use of antibodies or by tagging with fluorescent proteins, appeared as a great tool for such an aim. However, the spatial resolution of fluorescent microscopes is limited by the wavelength of light at around ~220 nm (Figure 1). While this resolution is sufficient to visualize cellular organelles, it is not sufficient to resolve the molecular organization of these complexes. In parallel with the development of the optical approaches, multiple tools have been invented to overcome this obstacle. For example, electron microscopy (EM) allows ultrastructural-level analyses of biological structures, and the detection of specific molecules can also be accomplished by immunogold labeling. However, achieving high-density immunogold labeling is challenging, and it is far from being efficient. Scanning probe and nearfield optical scanning microscopy also offer outstanding resolution, but, unfortunately, they are largely surface-bound and not able to image the intact cellular interior, and hence their popularity is limited. In the end, the main goal remained the same: to go under the diffraction limit by using visible light. The breakthrough came from revisiting the principles of fluorescence microscopy, and that is what we call fluorescence nanoscopy today (also often referred to as super-resolution microscopy (SRM)). By bridging the gap between conventional light microscopy and structural biology, the founders of these new concepts were awarded the Nobel Prize in Chemistry in 2014. In this review, we summarize the principles of the main nanoscopic approaches, and their impact on understanding the molecular organization of several highly conserved cellular machineries by using Drosophila models.
Figure 1. The resolution limit of optical microscopes. (A) In practice, even an extremely small point source of light (e.g., a single fluorophore) is still imaged as a spot of finite volume, known as the point spread function (PSF). Therefore, when two objects are close to each other, it is impossible to distinguish them. In order to discriminate them as two individual objects, they need to be separated by an adequate distance (d). This distance, by definition, is the resolution of the microscope. (B) The resolution of an optical system can be estimated by measuring the full-width at half-maximum (FWHM) of the PSF. (C) The spatial resolution of an optical imaging system is limited by the diffraction of the light, which was first described in 1873 by Ernst Abbe. This resolution limit in microscopy is calculated by the following equation: λ/2NA, where λ is the wavelength of the light, and NA is the numerical aperture of the objective. Accordingly, even in the most favorable condition, with a high NA objective and the lowest possible wavelength, the resolution will never be higher than ~220 nm.
3. Biological Insights Offered by Nanoscopy
As it is outlined above, despite the rather different rational designs, each SRM technique is suitable to break the resolution limit. Nevertheless, in large part owing to the differences in their technical concept, each method has its own benefits and drawbacks, which are summarized in
Table 1, together with the main technical features. The choice of method in the case of a biological sample of interest must take into account these differences, and ideally, it is advised to compare these approaches experimentally. For example, when we decided to study the sarcomeric distribution of a set of actin regulatory proteins, the
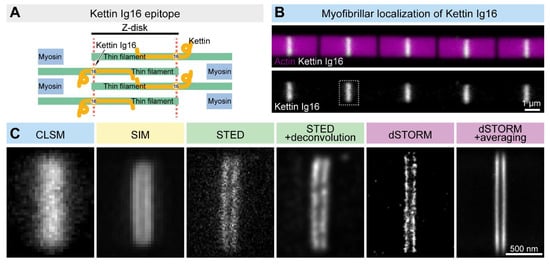
Drosophila Titin ortholog (Kettin) was used as a ‘resolution’ control. Based on immunogold EM images, the 16th immunoglobulin domain of Kettin (Ig16) is present in two stripes at the edges of the Z-disk separated by ~100 nm
[1]. Due to their close proximity, the individual ‘lines’ cannot be resolved as distinct structures with confocal laser scanning microscopes (
Figure 2). However, SIM or STED microscopy already provides significant improvements, while STORM is able to resolve the sarcomeric structures with a high resolution, which can be further improved by structure averaging (
Figure 2). Thus, Z-disk localization of Kettin not only perfectly demonstrated the resolving power of the different nanoscopic approaches but also provided us with a useful batch of information on the available techniques, which was key to our subsequent studies on sarcomere organization. This example will be reviewed, in detail, in the following sections together with several other
Drosophila models highlighting how resolution improvement can lead to major discoveries in various fields of life sciences.
Figure 2. Comparison of the lateral resolution of the different SRM approaches. (A) The Kettin Ig16 epitope is found in two stripes separated by ~100 nm in the Z-disk of IFM myofibrils. (B) Confocal imaging of the individual myofibrils reveals the Z-disk accumulation of the Kettin Ig16 epitope. The dotted rectangle marks the Z-disk presented in panel C. (C) The Kettin Ig16 signal at the Z-disk appears as a single band with confocal microscopy (CLSM), which can be resolved into two individual bands with various super-resolution approaches such as SIM, STED or dSTORM. dSTORM combined with structure averaging (on the right) can produce an image with the most outstanding resolution.
Table 1. Comparison of the different SRM techniques. *: low, **: medium, ***: high.
| |
SIM |
STED |
STORM/PALM |
ExM |
| Concept to overcome the resolution limit |
High-frequency information containing interference generated by patterned illumination |
Stimulated de-excitation is used to produce a narrower emission zone |
Fluorophores are modulated in the time scale to separate and localize them one by one |
Isotropic sample expansion is used to increase the distance between the molecules |
| Microscopy type |
Widefield |
Laser scanning confocal |
Widefield |
Widefield/laser scanning confocal/spinning disk confocal |
| Lateral resolution |
~100 nm (linear)
~50 nm (nonlinear) |
~30–50 nm |
~20 nm |
~70 nm
~25 nm (with SIM) |
| Axial resolution |
~300 nm (linear)
~120 nm (nonlinear) |
~30 nm |
~50 nm |
~200 nm
~60 nm (with SIM) |
| Fluorophore type |
Conventional fluorescent proteins and dyes |
Photostable dyes and fluorescent proteins |
STORM: photoswitchable dyes
PALM: photoswitchable fluorescent proteins/dyes |
Special labeling probe (able to survive the homogenization) |
| Phototoxicity |
*/** |
**/*** |
** |
- |
| Photobleaching |
**/*** |
**/*** |
* |
* |
| Live imaging |
Well suited |
Moderately suited |
Limitedly suited |
No |
| Post-image processing |
Yes |
No |
Yes |
Yes |
| Maximum number of simultaneous colors |
4 |
2 |
2–3 |
4 |
| Concerns |
Out-of-focus signals |
Photobleaching |
Over/under-labeling artifacts |
Time-consuming optimization |
 +1 credit
+1 credit