 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ramendra Pati Pandey | + 5025 word(s) | 5025 | 2021-08-04 08:38:18 | | | |
| 2 | Bruce Ren | -21 word(s) | 5004 | 2021-08-05 03:13:43 | | |
Video Upload Options
Among the several human fungal pathogens, Candida genus represents one of the most implicated in the clinical scenario. There exist several distinctive features that govern the establishment of Candida infections in addition to their capacity to adapt to multiple stress conditions inside humans which also include evasion of host immune responses. The complex fungal cell wall of the prevalent pathogen, Candida albicans, is one of the main targets of antifungal drugs and recognized by host immune cells. The wall consists of tiered arrangement of an outer thin but dense covering of mannan and inner buried layers of β-glucan and chitin. However, the pathogenic fungi adopt strategies to evade immune recognition by masking these molecules. This capacity to camouflage the immunogenic polysaccharide β-glucan from the host is a key virulence factor of C. albicans.
1. Introduction
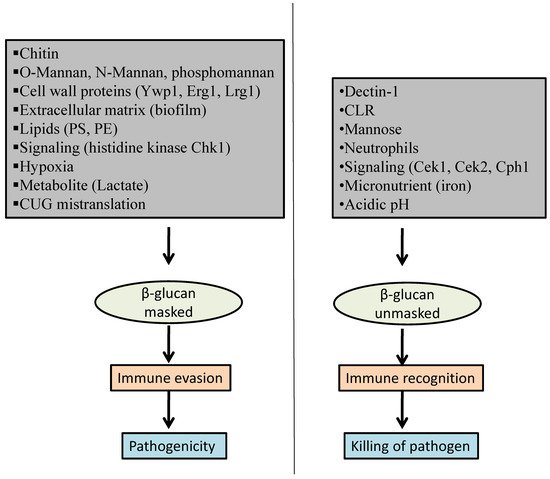
2. Factors That Influence β-Glucan Masking
2.1. Host-Pathogen Interaction
2.1.1. Dectin-1
2.1.2. C-Type Lectin Receptor (CLR)
3.1.3. Mannose Receptor
2.1.4. Neutrophil Extracellular Trap (NET)
2.2. Cell Wall Proteins
2.3. Chitin
2.4. O-Mannan, N-Mannan, and Phosphomannan
2.5. Extracellular Matrix
2.6. Lipids
2.7. Signaling Pathways
2.8. Micronutrient
2.9. Hypoxia
2.10. pH
2.11. Metabolites
2.12. Mistranslation of CUG
References
- Beardsley, J.; Halliday, C.L.; Chen, S.C.; Sorrell, T.C. Responding to the emergence of antifungal drug resistance: Perspectives from the bench and the bedside. Future Microbiol. 2018, 13, 1175–1191.
- Singh, S.; Fatima, Z.; Hameed, S. Predisposing factors endorsing Candida infections. Infez. Med. 2015, 23, 211–223.
- Perfect, J.R. The antifungal pipeline: A reality check. Nat. Rev. Drug Discov. 2017, 16, 603–616.
- Deorukhkar, S.C.; Saini, S.; Mathew, S. Virulence Factors Contributing to Pathogenicity of Candida tropicalis and Its Antifungal Susceptibility Profile. Int. J. Microbiol. 2014, 2014, 456878.
- Graus, M.S.; Wester, M.J.; Lowman, D.W.; Williams, D.L.; Kruppa, M.D.; Martinez, C.M.; Young, J.M.; Pappas, H.C.; Lidke, K.A.; Neumann, A.K. Mannan Molecular Substructures Control Nanoscale Glucan Exposure in Candida. Cell Rep. 2018, 24, 2432–2442.
- Goodridge, H.S.; Wolf, A.J.; Underhill, D.M. Beta-glucan recognition by the innate immune system. Immunol. Rev. 2009, 230, 38–50.
- Childers, D.S.; Avelar, G.M.; Bain, J.M.; Pradhan, A.; Larcombe, D.E.; Netea, M.G.; Erwig, L.P.; Gow, N.; Brown, A. Epitope Shaving Promotes Fungal Immune Evasion. mBio 2020, 11, e00984-20.
- Wester, M.J.; Lin, J.; Neumann, A.K. A computational model for regulation of nanoscale glucan exposure in Candida albicans. PLoS ONE 2017, 12, e0188599.
- Pradhan, A.; Avelar, G.M.; Bain, J.M.; Childers, D.; Pelletier, C.; Larcombe, D.E.; Shekhova, E.; Netea, M.G.; Brown, G.D.; Erwig, L.; et al. Non-canonical signalling mediates changes in fungal cell wall PAMPs that drive immune evasion. Nat. Commun. 2019, 10, 5315.
- Lin, J.; Wester, M.J.; Graus, M.S.; Lidke, K.A.; Neumann, A.K. Nanoscopic cell-wall architecture of an immunogenic ligand in Candida albicans during antifungal drug treatment. Mol. Biol. Cell 2016, 27, 1002–1014.
- Chen, T.; Jackson, J.W.; Tams, R.N.; Davis, S.E.; Sparer, T.E.; Reynolds, T.B. Exposure of Candida albicans β (1,3)-glucan is promoted by activation of the Cek1 pathway. PLoS Genet. 2019, 15, e1007892.
- Chen, S.M.; Zou, Z.; Qiu, X.R.; Hou, W.T.; Zhang, Y.; Fang, W.; Chen, Y.L.; Wang, Y.D.; Jiang, Y.Y.; Shen, H.; et al. The critical role of Dectin-1 in host controlling systemic Candida krusei infection. Am. J. Transl. Res. 2019, 11, 721–732.
- Granger, B.L. Propeptide genesis by Kex2-dependent cleavage of yeast wall protein 1 (Ywp1) of Candida albicans. PLoS ONE 2018, 13, e0207955.
- Granger, B.L. Accessibility and contribution to glucan masking of natural and genetically tagged versions of yeast wall protein 1 of Candida albicans. PLoS ONE 2018, 13, e0191194.
- Bain, J.M.; Louw, J.; Lewis, L.E.; Okai, B.; Walls, C.A.; Ballou, E.R.; Walker, L.A.; Reid, D.; Munro, C.A.; Brown, A.J.; et al. Candida albicans hypha formation and mannan masking of β-glucan inhibit macrophage phagosome maturation. mBio 2014, 5, e01874.
- Klippel, N.; Cui, S.; Groebe, L.; Bilitewski, U. Deletion of the Candida albicans histidine kinase gene CHK1 improves recognition by phagocytes through an increased exposure of cell wall beta-1,3-glucans. Microbiology 2010, 156, 3432–3444.
- Childers, D.S.; Avelar, G.M.; Bain, J.M.; Larcombe, D.E.; Pradhan, A.; Budge, S.; Heaney, H.; Brown, A. Impact of the Environment upon the Candida albicans Cell Wall and Resultant Effects upon Immune Surveillance. Curr. Top. Microbiol. Immunol. 2020, 425, 297–330.
- Hopke, A.; Brown, A.J.; Hall, R.A.; Wheeler, R.T. Dynamic fungal cell wall architecture in stress adaptation and immune evasion. Trends Microbiol. 2018, 26, 284–295.
- Marakalala, M.J.; Vautier, S.; Potrykus, J.; Walker, L.A.; Shepardson, K.M.; Hopke, A.; Mora-Montes, H.M.; Kerrigan, A.; Netea, M.G.; Murray, G.I.; et al. Differential adaptation of Candida albicans in vivo modulates immune recognition by dectin-1. PLoS Pathog. 2013, 9, e1003315.
- Brown, A.J.; Gow, N.A.; Warris, A.; Brown, G.D. Memory in fungal pathogens promotes immune evasion, colonisation, and infection. Trends Microbiol. 2019, 27, 219–230.
- Yadav, B.; Mora-Montes, H.M.; Wagener, J.; Cunningham, I.; West, L.; Haynes, K.; Brown, A.J.P.; Gow, N.A.R. Differences in fungal immune recognition by monocytes and macrophages: N-mannan can be a shield or activator of immune recognition. Cell Surf. 2020, 6, 100042.
- Cottier, F.; Hall, R.A. Face/off: The interchangeable side of Candida albicans. Front. Cell. Infect. Microbiol. 2020, 9, 471.
- Hall, R.A.; Bates, S.; Lenardon, M.D.; MacCallum, D.M.; Wagener, J.; Lowman, D.W.; Kruppa, M.D.; Williams, D.L.; Odds, F.C.; Brown, A.J.; et al. The Mnn2 mannosyltransferase family modulates mannoprotein fibril length, immune recognition and virulence of Candida albicans. PLoS Pathog. 2013, 9, e1003276.
- Lee, S.J.; Zheng, N.Y.; Clavijo, M.; Nussenzweig, M.C. Normal host defense during systemic candidiasis in mannose receptor-deficient mice. Infect. Immun. 2003, 71, 437–445.
- Zawrotniak, M.; Bochenska, O.; Karkowska-Kuleta, J.; Seweryn-Ozog, K.; Aoki, W.; Ueda, M.; Kozik, A.; Rapala-Kozik, M. Aspartic Proteases and Major Cell Wall Components in Candida albicans Trigger the Release of Neutrophil Extracellular Traps. Front. Cell Infect. Microbiol. 2017, 7, 414.
- Hopke, A.; Nicke, N.; Hidu, E.E.; Degani, G.; Popolo, L.; Wheeler, R.T. Neutrophil Attack Triggers Extracellular Trap-Dependent Candida Cell Wall Remodeling and Altered Immune Recognition. PLoS Pathog. 2016, 5, e1005644.
- Pericolini, E.; Perito, S.; Castagnoli, A.; Gabrielli, E.; Mencacci, A.; Blasi, E.; Vecchiarelli, A.; Wheeler, R.T. Epitope unmasking in vulvovaginal candidiasis is associated with hyphal growth and neutrophilic infiltration. PLoS ONE 2018, 13, e0201436.
- Levin, R.; Grinstein, S.; Canton, J. The life cycle of phagosomes: Formation, maturation, and resolution. Immunol. Rev. 2016, 273, 156–179.
- Wozniok, I.; Hornbach, A.; Schmitt, C.; Frosch, M.; Einsele, H.; Hube, B.; Löffler, J.; Kurzai, O. Induction of ERK-kinase signalling triggers morphotype-specific killing of Candida albicans filaments by human neutrophils. Cell. Microbiol. 2008, 10, 807–820.
- Seider, K.; Heyken, A.; Lüttich, A.; Miramón, P.; Hube, B. Interaction of pathogenic yeasts with phagocytes: Survival, persistence and escape. Curr. Opin. Microbiol. 2010, 13, 392–400.
- Small, A.G.; King, J.R.; Rathjen, D.A.; Ferrante, A. The role of phagocytes in immunity to Candida albicans. In Candida Albicans; IntechOpen: London, UK, 2018.
- Last, A.; Maurer, M.; Mosig, A.S.; Gresnigt, M.S.; Hube, B. In vitro infection models to study fungal-host interactions. FEMS Microbiol. Rev. 2021, 1, fuab005.
- Yang, M.; Solis, N.; Marshall, M.; Garleb, R.; Tingting, Z.; Wang, D.; Swidergall, M.; Pearlman, E.; Filler, S.; Liu, H. Control of β-glucan exposure by the endo-1,3-glucanase Eng1 in Candida albicans modulates virulence. bioRxiv 2020.
- Chen, T.; Wagner, A.S.; Tams, R.N.; Eyer, J.E.; Kauffman, S.J.; Gann, E.R.; Fernandez, E.J.; Reynolds, T.B. Lrg1 Regulates β (1,3)-Glucan Masking in Candida albicans through the Cek1 MAP Kinase Pathway. mBio 2019, 10, e01767-19.
- Qadota, H.; Python, C.P.; Inoue, S.B.; Arisawa, M.; Anraku, Y.; Zheng, Y.; Watanabe, T.; Levin, D.E.; Ohya, Y. Identification of yeast Rho1p GTPase as a regulatory subunit of 1,3-beta-glucan synthase. Science 1996, 272, 279–281.
- Fuller, R.S.; Brake, A.J.; Thorner, J. Intracellular targeting and structural conservation of a prohormone-processing endoprotease. Science 1989, 246, 482–486.
- McKenzie, C.G.; Koser, U.; Lewis, L.E.; Bain, J.M.; Mora-Montes, H.M.; Barker, R.N.; Gow, N.A.; Erwig, L.P. Contribution of Candida albicans cell wall components to recognition by and escape from murine macrophages. Infect. Immun. 2010, 78, 1650–1658.
- Gómez-Gaviria, M.; Lozoya-Pérez, N.E.; Staniszewska, M.; Franco, B.; Niño-Vega, G.A.; Mora-Montes, H.M. Loss of Kex2 Affects the Candida albicans Cell Wall and Interaction with Innate Immune Cells. J. Fungi 2020, 6, 57.
- Mora-Montes, H.M.; Netea, M.G.; Ferwerda, G.; Lenardon, M.D.; Brown, G.D.; Mistry, A.R.; Kullberg, B.J.; O’Callaghan, C.A.; Sheth, C.C.; Odds, F.C.; et al. Recognition and Blocking of Innate Immunity Cells by Candida albicans Chitin. Infect. Immun. 2011, 79, 1961–1970.
- Lee, K.K.; Maccallum, D.M.; Jacobsen, M.D.; Walker, L.A.; Odds, F.C.; Gow, N.A.; Munro, C.A. Elevated cell wall chitin in Candida albicans confers echinocandin resistance in vivo. Antimicrob. Agents Chemother. 2012, 56, 208–217.
- Gow, N.A.; Netea, M.G.; Munro, C.A.; Ferwerda, G.; Bates, S.; Mora-Montes, H.M.; Walker, L. Immune recognition of Candida albicans beta-glucan by dectin-1. J. Infect. Dis. 2007, 196, 1565–1571.
- Martínez-Duncker, I.; Díaz-Jímenez, D.F.; Mora-Montes, H.M. Comparative Analysis of Protein Glycosylation Pathways in Humans and the Fungal Pathogen Candida albicans. Int. J. Microbiol. 2014, 2014, 1–16.
- Netea, M.G.; Gow, N.A.; Munro, C.A.; Bates, S.; Collins, C.; Ferwerda, G.; Hobson, R.P.; Bertram, G.; Hughes, H.B.; Jansen, T.; et al. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J. Clin. Investig. 2006, 116, 1642–1650.
- Cambi, A.; Netea, M.G.; Mora-Montes, H.M.; Gow, N.A.; Hato, S.V.; Lowman, D.W.; Kullberg, B.J.; Torensma, R.; Williams, D.L.; Figdor, C.G. Dendritic cell interaction with Candida albicans critically depends on N-linked mannan. J. Biol. Chem. 2008, 283, 20590–20599.
- Zarnowski, R.; Westler, W.M.; Lacmbouh, G.A.; Marita, J.M.; Bothe, J.R.; Bernhardt, J.; Lounes-Hadj Sahraoui, A.; Fontaine, J.; Sanchez, H.; Hatfield, R.D.; et al. Novel entries in a fungal biofilm matrix encyclopedia. mBio 2014, 5, e01333-14.
- Sheppard, D.C.; Howell, P.L. Biofilm Exopolysaccharides of Pathogenic Fungi: Lessons from Bacteria. J. Biol. Chem. 2016, 291, 12529–12537.
- Chen, Y.L.; Montedonico, A.E.; Kauffman, S.; Dunlap, J.R.; Menn, F.M.; Reynolds, T.B. Phosphatidylserine synthase and phosphatidylserine decarboxylase are essential for cell wall integrity and virulence in Candida albicans. Mol. Microbiol. 2010, 75, 1112–1132.
- Monge, R.A.; Román, E.; Nombela, C.; Pla, J. The MAP kinase signal transduction network in Candida albicans. Microbiology 2006, 152, 905–912.
- Dagley, M.J.; Gentle, I.E.; Beilharz, T.H.; Pettolino, F.A.; Djordjevic, J.T.; Lo, T.L.; Uwamahoro, N.; Rupasinghe, T.; Tull, D.L.; McConville, M.; et al. Cell wall integrity is linked to mitochondria and phospholipid homeostasis in Candida albicans through the activity of the post-transcriptional regulator Ccr4-Pop2. Mol. Microbiol. 2011, 79, 968–989.
- Davis, S.E.; Hopke, A.; Minkin, S.C., Jr.; Montedonico, A.E.; Wheeler, R.T.; Reynolds, T.B. Masking of β(1-3)-glucan in the cell wall of Candida albicans from detection by innate immune cells depends on phosphatidylserine. Infect. Immun. 2014, 82, 4405–4413.
- Galán-Díez, M.; Arana, D.M.; Serrano-Gómez, D.; Kremer, L.; Casasnovas, J.M.; Ortega, M.; Cuesta-Domínguez, A.; Corbí, A.L.; Pla, J.; Fernández-Ruiz, E. Candida albicans beta-glucan exposure is controlled by the fungal CEK1-mediated mitogen-activated protein kinase pathway that modulates immune responses triggered through dectin-1. Infect. Immun. 2010, 78, 1426–1436.
- Roman, E.; Correia, I.; Salazin, A.; Fradin, C.; Jouault, T.; Poulain, D.; Liu, F.T.; Pla, J. The Cek1-mediated MAP kinase pathway regulates exposure of α-(1,2) and β-(1,2)-mannosides in the cell wall of Candida albicans modulating immune recognition. Virulence 2016, 7, 558–577.
- Correia, I.; Prieto, D.; Román, E.; Wilson, D.; Hube, B.; Alonso-Monge, R.; Pla, J. Cooperative Role of MAPK Pathways in the Interaction of Candida albicans with the Host Epithelium. Microorganisms 2019, 8, 48.
- Kruppa, M.; Goins, T.; Cutler, J.E.; Lowman, D.; Williams, D.; Chauhan, N.; Menon, V.; Singh, P.; Li, D.; Calderone, R. The role of the Candida albicans histidine kinase [CHK1) gene in the regulation of cell wall mannan and glucan biosynthesis. FEMS Yeast Res. 2003, 3, 289–299.
- Kruppa, M.; Jabra-Rizk, M.A.; Meiller, T.F.; Calderone, R. The histidine kinases of Candida albicans: Regulation of cell wall mannan biosynthesis. FEMS Yeast Res. 2004, 4, 409–416.
- Ramanan, N.; Wang, Y. A high-affinity iron permease essential for Candida albicans virulence. Science 2000, 288, 1062–1064.
- Almeida, R.S.; Wilson, D.; Hube, B. Candida albicans iron acquisition within the host. FEMS Yeast Res. 2009, 9, 1000–1012.
- Potrykus, J.; Stead, D.; Maccallum, D.M.; Urgast, D.S.; Raab, A.; van Rooijen, N.; Feldmann, J.; Brown, A.J. Fungal iron availability during deep seated candidiasis is defined by a complex interplay involving systemic and local events. PLoS Pathog. 2013, 9, e1003676.
- Grahl, N.; Demers, E.G.; Lindsay, A.K.; Harty, C.E.; Willger, S.D.; Piispanen, A.E.; Hogan, D.A. Mitochondrial activity and Cyr1 are key regulators of Ras1 activation of C. albicans virulence pathways. PLoS Pathog. 2015, 11, e1005133.
- Finch, C.A.; Gollnick, P.D.; Hlastala, M.P.; Miller, L.R.; Dillmann, E.; Mackler, B. Lactic acidosis as a result of iron deficiency. J. Clin. Investig. 1979, 64, 129–137.
- Amarsaikhan, N.; Sands, E.M.; Shah, A.; Abdolrasouli, A.; Reed, A.; Slaven, J.E.; Armstrong-James, D.; Templeton, S.P. Caspofungin Increases Fungal Chitin and Eosinophil and γδ T Cell-Dependent Pathology in Invasive Aspergillosis. J. Immunol. 2017, 199, 624–632.
- Lamaris, G.A.; Lewis, R.E.; Chamilos, G.; May, G.S.; Safdar, A.; Walsh, T.J.; Raad, I.I.; Kontoyiannis, D.P. Caspofungin-mediated beta-glucan unmasking and enhancement of human polymorphonuclear neutrophil activity against Aspergillus and non-Aspergillus hyphae. J. Infect. Dis. 2008, 198, 186–192.
- Grahl, N.; Shepardson, K.M.; Chung, D.; Cramer, R.A. Hypoxia and fungal pathogenesis: To air or not to air? Eukaryot. Cell 2012, 11, 560–570.
- Synnott, J.M.; Guida, A.; Mulhern-Haughey, S.; Higgins, D.G.; Butler, G. Regulation of the hypoxic response in Candida albicans. Eukaryot. Cell 2010, 9, 1734–1746.
- Sellam, A.; van Het Hoog, M.; Tebbji, F.; Beaurepaire, C.; Whiteway, M.; Nantel, A. Modeling the transcriptional regulatory network that controls the early hypoxic response in Candida albicans. Eukaryot. Cell 2014, 13, 675–690.
- Stichternoth, C.; Ernst, J.F. Hypoxic adaptation by Efg1 regulates biofilm formation by Candida albicans. Appl. Environ. Microbiol. 2009, 75, 3663–3672.
- Hickman, M.J.; Spatt, D.; Winston, F. The Hog1 mitogen-activated protein kinase mediates a hypoxic response in Saccharomyces cerevisiae. Genetics 2011, 188, 325–338.
- Lopes, J.P.; Stylianou, M.; Backman, E.; Holmberg, S.; Jass, J.; Claesson, R.; Urban, C.F. Evasion of Immune Surveillance in Low Oxygen Environments Enhances Candida albicans Virulence. mBio 2018, 9, e02120-e18.
- Pradhan, A.; Avelar, G.M.; Bain, J.M.; Childers, D.S.; Larcombe, D.E.; Netea, M.G.; Shekhova, E.; Munro, C.A.; Brown, G.D.; Erwig, L.P.; et al. Hypoxia Promotes Immune Evasion by Triggering β-Glucan Masking on the Candida albicans Cell Surface via Mitochondrial and cAMP-Protein Kinase a Signaling. mBio 2018, 9, e01318-e18.
- Linhares, I.M.; Summers, P.R.; Larsen, B.; Giraldo, P.C.; Witkin, S.S. Contemporary perspectives on vaginal pH and lactobacilli. Am. J. Obstet. Gynecol. 2011, 204, 120.e1–120.e5.
- Sherrington, S.L.; Sorsby, E.; Mahtey, N.; Kumwenda, P.; Lenardon, M.D.; Brown, I.; Ballou, E.R.; MacCallum, D.M.; Hall, R.A. Adaptation of Candida albicans to environmental pH induces cell wall remodelling and enhances innate immune recognition. PLoS Pathog. 2017, 13, e1006403.
- Wheeler, R.T.; Kombe, D.; Agarwala, S.D.; Fink, G.R. Dynamic, morphotype-specific Candida albicans beta-glucan exposure during infection and drug treatment. PLoS Pathog. 2008, 4, e1000227.
- Ene, I.V.; Adya, A.K.; Wehmeier, S.; Brand, A.C.; MacCallum, D.M.; Gow, N.A.; Brown, A.J. Host carbon sources modulate cell wall architecture, drug resistance and virulence in a fungal pathogen. Cell Microbiol. 2012, 14, 1319–1335.
- Ene, I.V.; Walker, L.A.; Schiavone, M.; Lee, K.K.; Martin-Yken, H.; Dague, E.; Gow, N.A.; Munro, C.A.; Brown, A.J. Cell wall remodeling enzymes modulate fungal cell wall elasticity and osmotic stress resistance. mBio 2015, 6, e00986.
- Ballou, E.R.; Avelar, G.M.; Childers, D.S.; Mackie, J.; Bain, J.M.; Wagener, J.; Kastora, S.L.; Panea, M.D.; Hardison, S.E.; Walker, L.A. Lactate signalling regulates fungal β-glucan masking and immune evasion. Nat. Microbiol. 2016, 2, 16238.
- Vieira, N.; Casal, M.; Johansson, B.; MacCallum, D.M.; Brown, A.J.; Paiva, S. Functional specialization and differential regulation of short-chain carboxylic acid transporters in the pathogen Candida albicans. Mol. Microbiol. 2010, 75, 1337–1354.
- Maidan, M.M.; De Rop, L.; Serneels, J.; Exler, S.; Rupp, S.; Tournu, H.; Thevelein, J.M.; Van Dijck, P. The G protein-coupled receptor Gpr1 and the Galpha protein Gpa2 act through the cAMP-protein kinase A pathway to induce morphogenesis in Candida albicans. Mol. Biol. Cell 2005, 16, 1971–1986.
- Karababa, M.; Valentino, E.; Pardini, G.; Coste, A.T.; Bille, J.; Sanglard, D. CRZ1, a target of the calcineurin pathway in Candida albicans. Mol. Microbiol. 2006, 59, 1429–1451.
- Miranda, I.; Silva-Dias, A.; Rocha, R.; Teixeira-Santos, R.; Coelho, C.; Gonçalves, T.; Santos, M.A.; Pina-Vaz, C.; Solis, N.V.; Filler, S.G.; et al. Candida albicans CUG mistranslation is a mechanism to create cell surface variation. mBio 2013, 4, e00285-e13.
- Miranda, I.; Rocha, R.; Santos, M.C.; Mateus, D.D.; Moura, G.R.; Carreto, L.; Santos, M.A. A genetic code alteration is a phenotype diversity generator in the human pathogen Candida albicans. PLoS ONE 2007, 2, e996.
- Gomes, A.C.; Miranda, I.; Silva, R.M.; Moura, G.R.; Thomas, B.; Akoulitchev, A.; Santos, M.A. A genetic code alteration generates a proteome of high diversity in the human pathogen Candida albicans. Genome Biol. 2007, 8, R206.
- Almeida, R.S.; Brunke, S.; Albrecht, A.; Thewes, S.; Laue, M.; Edwards, J.E.; Filler, S.G.; Hube, B. The hyphal-associated adhesin and invasin Als3 of Candida albicans mediates iron acquisition from host ferritin. PLoS Pathog. 2008, 4, e1000217.
- Rauceo, J.M.; Gaur, N.K.; Lee, K.G.; Edwards, J.E.; Klotz, S.A.; Lipke, P.N. Global cell surface conformational shift mediated by a Candida albicans adhesin. Infect. Immun. 2004, 72, 4948–4955.


