 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Strahil Strashilov | + 2405 word(s) | 2405 | 2021-07-23 05:12:56 | | | |
| 2 | Catherine Yang | Meta information modification | 2405 | 2021-08-05 04:36:39 | | | | |
| 3 | Conner Chen | Meta information modification | 2405 | 2021-09-22 04:37:55 | | |
Video Upload Options
Melanoma develops from malignant transformations of the pigment-producing melanocytes. If located in the basal layer of the skin epidermis, melanoma is referred to as cutaneous, which is more frequent. However, as melanocytes are be found in the eyes, ears, gastrointestinal tract, genitalia, urinary system, and meninges, cases of mucosal melanoma or other types (e.g., ocular) may occur. The incidence and morbidity of cutaneous melanoma (cM) are constantly increasing worldwide.
1. Introduction
The term melanoma was first used in 1812 by René Laennec to describe a case of metastatic dissemination of the disease [1]. Cutaneous melanoma (cM) develops from malignant transformations of pigment-producing melanocytes in the basal layer of the skin epidermis. Non-cutaneous melanoma arises from malignantly transformed melanocytes in the uvea [2][3], gastrointestinal tract [4][5], genitalia [6][7], urinary system [8][9], meninges [10][11], etc. The incidence and morbidity of cM are constantly increasing worldwide. Australia and New Zealand are world leaders in this regard with a morbidity rate of 54/100,000 and a mortality rate of 5.6/100,000 for 2015 [12].
2. Pathogenesis of Cutaneous Melanoma
2.1. MAPK Pathway
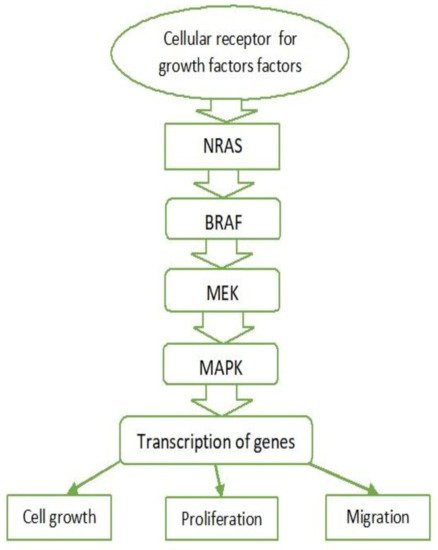
2.2. BRAF
2.3. RAS
2.4. c-KIT
2.5. c-MET and HGF
2.6. Other Factors
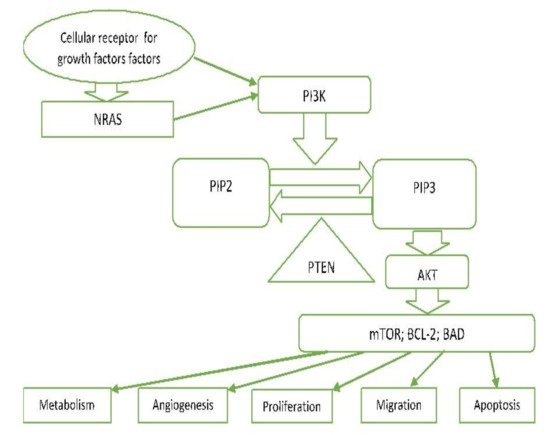
2.7. PI3K/PTEN/AKT Pathway (Phosphatidylinositol 3-kinase Pathway)
PI3K-AKT is another critical signalling pathway in the cell. It is involved in the regulation of cell survival, growth, and apoptosis [13][23] (Figure 2).
2.8. MITF Signalling
2.9. p53
2.10. Hypoxia-Induced Factor (HIF)
2.11. Notch Signalling Pathway
3.12. Other Factors Important in the Pathogenesis of Melanoma
References
- Chin, L.; Merlino, G.; Depinho, R.A. Malignant melanoma: Modern black plague and genetic black box. Genes Dev. 1998, 12, 3467–3481.
- Sayan, M.; Mamidanna, S.; Oncel, D.; Jan, I.; Vergalasova, I.; Weiner, J.; Ohri, N.; Acikalin, B.; Chundury, A. Clinical management of uveal melanoma: A comprehensive review with a treatment algorithm. Radiat. Oncol. J. 2020, 38, 162–169.
- Jager, M.J.; Shields, C.L.; Cebulla, C.M.; Abdel-Rahman, M.H.; Grossniklaus, H.E.; Stern, M.H.; Carvajal, R.D.; Belfort, R.N.; Jia, R.; Shields, J.A.; et al. Uveal melanoma. Nat. Rev. Dis. Primers 2020, 6, 1–25.
- Zheng, Y.; Cong, C.; Su, C.; Sun, Y.; Xing, L. Epidemiology and survival outcomes of primary gastrointestinal melanoma: A SEER-based population study. Int. J. Clin. Oncol. 2020, 25, 1951–1959.
- Kahl, A.R.; Gao, X.; Chioreso, C.; Goffredo, P.; Hassan, I.; Charlton, M.E.; Lin, C. Presentation, Management, and Prognosis of Primary Gastrointestinal Melanoma: A Population-Based Study. J. Surg. Res. 2021, 260, 46–55.
- Wohlmuth, C.; Wohlmuth-Wieser, I.; May, T.; Vicus, D.; Gien, L.T.; LaFramboise, S. Malignant Melanoma of the Vulva and Vagina: A US Population-Based Study of 1863 Patients. Am. J. Clin. Dermatol. 2019, 21, 285–295.
- Sun, X.; Gu, Y.; Xie, J.; Wang, L.; Zhou, Q. Melanoma of female genital tract: A clinicopathological analysis of 5 cases. Zhonghua Bing Li Xue Za Zhi 2020, 49, 834–836.
- Acikalin, A.; Bagir, E.; Karim, S.; Bisgin, A.; Izol, V.; Erdogan, S. Primary melanoma of the urinary tract; Clinicopathologic and molecular review of a case series. Pathol. Res. Pract. 2020, 216, 153095.
- Kaboré, F.A.; Ouédraogo, B.; Ido, F.A.H.A.; Hafing, T.; Karama, H.; Traoré, O. Primary malignant melanoma of the urethra in women: About a case. Urol. Case Rep. 2021, 35, 101542.
- Machado, A.K.L.P.; Nunes, D.B.C.; Carneiro, F.R.O.; Mendes, A.M.D. Primary melanoma of leptomeninge in a patient with giant congenital melanocytic nevus. An. Bras. Dermatol. 2020, 95, 404–406.
- Cao, Y.; Wang, Y.-B.; Tan, X.-Y.; Cui, Y.-H.; Zhao, G. Multifocal primary amelanotic meningeal melanomas mimicking lymphoma: A case report and literature review. Br. J. Neurosurg. 2020, 15, 1–5.
- Karimkhani, C.; Green, A.; Nijsten, T.; Weinstock, M.; Dellavalle, R.; Naghavi, M.; Fitzmaurice, C. The global burden of melanoma: Results from the Global Burden of Disease Study 2015. Br. J. Dermatol. 2017, 177, 134–140.
- Garbe, C.; Bauer, J. Melanoma. In Dermatology, 3rd ed.; Bolognia, J., Jorizzo, J., Schaffer, J., Eds.; Elsevier: Amsterdam, The Netherlands; Saunders: Philadelphia, PA, USA, 2012; Volume 2, pp. 1885–1914.
- Hima, P.; Yacoub, N.; Mishra, R.; White, A.; Long, Y.; Alanazi, S.; Garrett, J.T. Current Advances in the Treatment of BRAF-Mutant Melanoma. Cancers 2020, 12, 482.
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghì, A.; Spandidos, D.A.; Mccubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy. Int. J. Oncol. 2018, 52, 1071–1080.
- Lo, J.; Fisher, D. Melanoma pathogenesis. In BRAF Targets in Melanoma; Sullivan, R., Ed.; Springer: New York, NY, USA, 2015; pp. 25–45.
- Davies, M.; Garraway, L. Molecular biology of cutaneous melanoma. In Principles and Practice of Oncology; Devita, V., Hellman, T., Rosenberg, S., Eds.; Wolters Kluwer: Philadelphia, PA, USA, 2015; pp. 1337–1345.
- Sullivan, R.; Fisher, D. The Molecular Biology of Melanoma 2017. Available online: https://www.uptodate.com/contents/ (accessed on 12 June 2021).
- Kiuru, M.; Busam, K.J. The NF1 gene in tumor syndromes and melanoma. Lab. Investig. 2017, 97, 146–157.
- Meng, D.; Carvajal, R.D. KIT as an Oncogenic Driver in Melanoma: An Update on Clinical Development. Am. J. Clin. Dermatol. 2019, 20, 315–323.
- Czyz, M. HGF/c-MET Signaling in Melanocytes and Melanoma. Int. J. Mol. Sci. 2018, 19, 3844.
- Williams, E.A.; Montesion, M.; Shah, N.; Sharaf, R.; Pavlick, D.C.; Sokol, E.S.; Alexander, B.; Venstrom, J.; Elvin, J.A.; Ross, J.S.; et al. Melanoma with in-frame deletion of MAP2K1: A distinct molecular subtype of cutaneous melanoma mutually exclusive from BRAF, NRAS, and NF1 mutations. Mod. Pathol. 2020, 33, 2397–2406.
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; McArthur, G.; Ribas, A.; et al. Melanoma. Nat. Rev. Dis. Primers 2015, 1, 15003.
- Roesch, A.; Volkenandt, M. Melanoma. In Dermatology, 3rd ed.; Braun-Falco, O., Plewig, G., Wolf, H., Burgdorf, W.H.C., Eds.; Springer: Berlin, Germany, 2009; pp. 1416–1432.
- Ballesteros-Álvarez, J.; Dilshat, R.; Fock, V.; Möller, K.; Karl, L.; Larue, L.; Ögmundsdóttir, M.H.; Steingrímsson, E. MITF and TFEB cross-regulation in melanoma cells. PLoS ONE 2020, 15, e0238546.
- Garraway, L.A.; Widlund, H.; Rubin, M.A.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, J.D.A.; Granter, S.R.; Du, J.; et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nat. Cell Biol. 2005, 436, 117–122.
- Ugurel, S.; Houben, R.; Schrama, D.; Voigt, H.; Zapatka, M.; Schadendorf, D.; Bröcker, E.B.; Becker, J.C. Microphthalmia-Associated Transcription Factor Gene Amplification in Metastatic Melanoma Is a Prognostic Marker for Patient Survival, But Not a Predictive Marker for Chemosensitivity and Chemotherapy Response. Clin. Cancer Res. 2007, 13, 6344–6350.
- Du, J.; Widlund, H.; Horstmann, M.A.; Ramaswamy, S.; Ross, K.; Huber, W.E.; Nishimura, E.K.; Golub, T.R.; Fisher, D.E. Critical role of CDK2 for melanoma growth linked to its melanocyte-specific transcriptional regulation by MITF. Cancer Cell 2004, 6, 565–576.
- Levy, C.; Khaled, M.; Fisher, D.E. MITF: Master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 2006, 12, 406–414.
- Sherr, C.J. Principles of Tumor Suppression. Cell 2004, 116, 235–246.
- Avery-Kiejda, K.A.; Bowden, N.A.; Croft, A.J.; Scurr, L.L.; Kairupan, C.F.; Ashton, K.A.; Talseth-Palmer, B.A.; Rizos, H.; Zhang, X.D.; Scott, R.J.; et al. P53 in human melanoma fails to regulate target genes associated with apoptosis and the cell cycle and may contribute to proliferation. BMC Cancer 2011, 11, 1–17.
- Sirigu, P.; Piras, F.; Minerba, L.; Murtas, D.; Maxia, C.; Colombari, R.; Corbu, A.; Perra, M.T.; Ugalde, J. Prognostic prediction of the immunohistochemical expression of p16 and p53 in cutaneous melanoma: A comparison of two populations from different geographical regions. Eur. J. Histochem. 2006, 50, 191–198.
- Ragnarsson-Olding, B.; Platz, A.; Olding, L.; Ringborg, U. p53 protein expression and TP53 mutations in malignant melanomas of sun-sheltered mucosal membranes versus chronically sun-exposed skin. Melanoma Res. 2004, 14, 395–401.
- Smalley, K.S.; Contractor, R.; Haass, N.K.; Kulp, A.N.; Atilla-Gokcumen, G.E.; Williams, U.S.; Bregman, H.; Flaherty, K.T.; Soengas, M.S.; Meggers, E.; et al. An Organometallic Protein Kinase Inhibitor Pharmacologically Activates p53 and Induces Apoptosis in Human Melanoma Cells. Cancer Res. 2007, 67, 209–217.
- Onder, T.; Gupta, P.B.; Mani, S.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of E-Cadherin Promotes Metastasis via Multiple Downstream Transcriptional Pathways. Cancer Res. 2008, 68, 3645–3654.
- Wouters, B.G.; Koritzinsky, M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat. Rev. Cancer 2008, 8, 851–864.
- Bachmann, I.; Ladstein, R.; Straume, O.; Naumov, G.; Akslen, L. Tumor necrosis is associated with increased alphavbeta3 integrin expression and poor prognosis in nodular cutaneous melanomas. BMC Cancer 2008, 8, 1–10.
- Chang, S.-H.; Worley, L.A.; Onken, M.; Harbour, J.W. Prognostic biomarkers in uveal melanoma: Evidence for a stem cell-like phenotype associated with metastasis. Melanoma Res. 2008, 18, 191–200.
- Hurlbut, G.D.; Kankel, M.W.; Lake, R.J.; Artavanis-Tsakonas, S. Crossing paths with Notch in the hyper-network. Curr. Opin. Cell Biol. 2007, 19, 166–175.
- Balint, K.; Xiao, M.; Pinnix, C.C.; Soma, A.; Veres, I.; Juhasz, I.; Brown, E.J.; Capobianco, A.J.; Herlyn, M.; Liu, Z.-J. Activation of Notch1 signaling is required for beta-catenin-mediated human primary melanoma progression. J. Clin. Investig. 2005, 115, 3166–3176.
- Hoek, K.; Rimm, D.L.; Williams, K.R.; Zhao, H.; Ariyan, S.; Lin, A.; Kluger, H.M.; Berger, A.J.; Cheng, E.; Trombetta, E.S.; et al. Expression Profiling Reveals Novel Pathways in the Transformation of Melanocytes to Melanomas. Cancer Res. 2004, 64, 5270–5282.
- Pinnix, C.C.; Lee, J.T.; Liu, Z.-J.; McDaid, R.; Balint, K.; Beverly, L.J.; Brafford, P.A.; Xiao, M.; Himes, B.; Zabierowski, S.E.; et al. Active Notch1 Confers a Transformed Phenotype to Primary Human Melanocytes. Cancer Res. 2009, 69, 5312–5320.
- Bedogni, B.; Warneke, J.A.; Nickoloff, B.J.; Giaccia, A.J.; Powell, M.B. Notch1 is an effector of Akt and hypoxia in melanoma development. J. Clin. Investig. 2008, 118, 3660–3670.
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.P.; Pincus, L.B.; Ruben, B.S.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936.
- Konsoulova, A. Principles of cancer immunobiology and immunotherapy of solid tumors. In Immunopathology and Immunomodulation; Metodiev, K., Ed.; 2015; pp. 77–100.
- Schadendorf, D.; Kochs, C.; Livingstone, E. Introduction to cutaneous melanoma. In Handbook of Cutaneous Melanoma—A Guide to Diagnosis and Treatment; Schadendorf, D., Kochs, C., Livingstone, E., Eds.; Springer Healthcare: New York, NY, USA, 2013; pp. 1–12.
- Lugović, L.; Situm, M.; Kos, L. Malignant melanoma--future prospects. Acta Dermatovenerol. Croat. ADC 2005, 13, 36–43.
- Sullivan, R.J.; Atkins, M.B.; Kirkwood, J.M.; Agarwala, S.S.; Clark, J.I.; Ernstoff, M.S.; Fecher, L.; Gajewski, T.F.; Gastman, B.; Lawson, D.H.; et al. An update on the Society for Immunotherapy of Cancer consensus statement on tumor immunotherapy for the treatment of cutaneous melanoma: Version 2.0. J. Immunother. Cancer 2018, 6, 1–23.
- Weiss, S.A.; Wolchok, J.D.; Sznol, M. Immunotherapy of Melanoma: Facts and Hopes. Clin. Cancer Res. 2019, 25, 5191–5201.
- Lade-Keller, J.; Riber-Hansen, R.; Guldberg, P.; Schmidt, H.; Hamilton-Dutoit, S.; Steiniche, T. E- to N-cadherin switch in melanoma is associated with decreased expression of phosphatase and tensin homolog and cancer progression. Br. J. Dermatol. 2013, 169, 618–628.


