Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Paul J.F. Rider | + 3661 word(s) | 3661 | 2021-07-20 10:30:00 | | | |
| 2 | Camila Xu | Meta information modification | 3661 | 2021-08-04 08:03:12 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Rider, P.J. Tumor Microenvironment and Oncolytic Virotherapy. Encyclopedia. Available online: https://encyclopedia.pub/entry/12728 (accessed on 07 February 2026).
Rider PJ. Tumor Microenvironment and Oncolytic Virotherapy. Encyclopedia. Available at: https://encyclopedia.pub/entry/12728. Accessed February 07, 2026.
Rider, Paul J.f.. "Tumor Microenvironment and Oncolytic Virotherapy" Encyclopedia, https://encyclopedia.pub/entry/12728 (accessed February 07, 2026).
Rider, P.J. (2021, August 03). Tumor Microenvironment and Oncolytic Virotherapy. In Encyclopedia. https://encyclopedia.pub/entry/12728
Rider, Paul J.f.. "Tumor Microenvironment and Oncolytic Virotherapy." Encyclopedia. Web. 03 August, 2021.
Copy Citation
The Tumor Microenvironment (TME) represents an important barrier that can hinder immune responses against tumors and also attenuate immunotherapeutic efficacy.
herpes
VC2
immunotherapy
oncolytic virus
herpesvirus
HSV
1. Tumor Microenvironment
The TME represents an important barrier that can hinder immune responses against tumors and also attenuate immunotherapeutic efficacy (Figure 1). In situ immune components of tumors, including T cells, B cells, natural killer cells (NK), dendritic cells (DCs), macrophages, granulocytes as well as suppressive regulatory cells subtypes such as regulatory T cells (Tregs), regulatory B cells (Bregs), and myeloid-derived suppressor cells (MDSCs), play crucial roles in the host cancer immunosurveillance system, although their contribution varies between tumor types and among patients with the same cancer [1]. The type, density, and location of immune infiltrates within a tumor have been suggested to have a clinical prognostic impact [2]. Importantly, the infiltration of T cells, particularly CD8+ T cells, have has been shown to correlate with augmented responses to immunotherapy and improved survival [3][4][5].
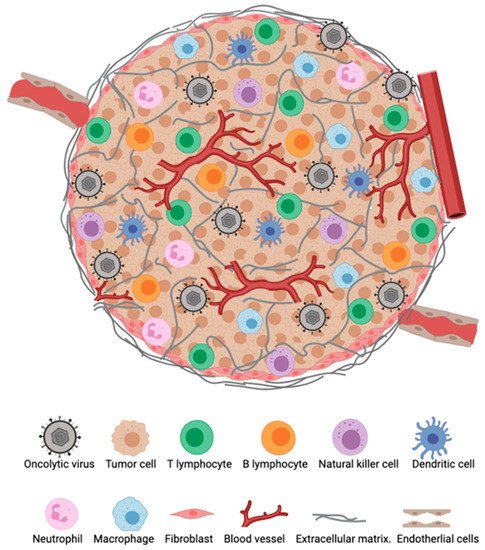
Figure 1. The tumor microenvironment (TME). Major cellular constituents of the TME during oncolytic virotherapy (OVT).
Tumors can be classified as immunologically “hot” or “cold” [4][6]. “Hot” or inflamed tumors have an abundance of infiltrating T cells and molecular signatures of immune activation and are usually responsive to immunotherapy [1]. By contrast, “cold” tumors are defined to have limited number of infiltrating T cells and are not sufficiently primed for immune recognition [1]. These cold tumors often contain Tregs, Bregs, and MDSCs, which prevent cytotoxic immune cells from penetrating into the TME [1]. Therefore, turning a cold tumor hot is essential and may significantly enhance the therapeutic efficacy of cancer immunotherapies. Unfortunately, only a minority of patients with hot cancers seem to respond and benefit from immunotherapies like checkpoint blockers, cancer vaccines and CAR-T cells. To improve clinical outcomes in patients with a variety of cancer types, significant efforts are currently undertaken to combine immunotherapies themselves or with traditional cancer therapies, such as chemotherapy, radiation therapy, or surgery [4].
The generation of an anti-tumor response involves a cyclic process referred to as the cancer-immunity cycle [7]. This cycle begins with liberation of neoantigens generated as a result of spontaneous immunogenic cell death, chemotherapy, radiation, and/or OVT among others. The released neoantigens are captured and processed by antigen-presenting cells (APCs), which migrate to the draining lymph nodes to prime and activate tumor-specific T cells. Tumor-specific T cells traffic to and infiltrate the tumor bed where they recognize and kill cancer cells. The killing of cancer cells releases additional tumor antigens, fueling the cancer immunity cycle.
2. Tumor Microenvironment and Oncolytic Virotherapy
2.1. oHSV and Anti-Tumor Immune Responses
A number of candidate oHSVs are currently in development [8]. Our group and others have demonstrated the significant impact of oHSV OVT on the development of anti-tumor immune responses in pre-clinical or clinical studies [9][10][11][12]. Specifically, these studies have highlighted the ability of oHSVs to change the TME from “cold” or immunosuppressive to “hot” or immunostimulatory as a component of their efficacy. Given the central role of the TME in controlling the development of anti-tumor immune responses, understanding the cellular and molecular changes in the TME during OVT would inform the development of more efficacious immunotherapies for the treatment of human and animal cancers. While the pre-clinical pipeline of oHSV is rich with promising candidates [9][10][11][12][13], we focus in this section on several oHSVs for which there is clinical data.
T-VEC™ is the first FDA approved OVT for treatment of patients with unresectable melanoma [14]. T-VEC is a modified HSV-1 OV derived from the JS-1 strain in which two copies of its ICP34.5 genes which encodes the neurovirulence factor are deleted [15]. The ICP34.5 gene is also important for viral replication, viral egress from cells and prevention of cellular block to viral protein synthesis in infected cells through its inhibitory activity on the protein kinase R (PKR) pathway [16][17][18]. PKR stops protein synthesis by phosphorylating the eukaryotic translation initiation factor 2 alpha (eIF2α) [18]. The ICP34.5 protein redirects protein phosphatase 1α (PP1α), which reverses the phosphorylation of eIF2α to restore protein synthesis [18]. Deletion of the ICP34.5 gene decreases pathogenesis and prevents replication of the virus in normal cells. In addition, T-VEC contains a deletion in the gene encoding the ICP47 protein [15]. HSV-1 uses the ICP47 protein to inhibit transporter associated with antigen presentation (TAP), thus it downregulates antigen presentation through MHC Class I, as a way of evading immune response during infection [19]. The deletion of the ICP47 gene increases antigen presentation and enhances viral immunogenicity. Additionally, ICP47 inactivation in T-VEC puts the herpes unique short 11 (US11) under immediate early promoter control which results in enhanced and earlier expression of US11, whose gene product inhibits PKR activity [20]. Finally, two copies of the human granulocyte–macrophage colony stimulating factor (GM-CSF) gene are inserted in place of the deleted ICP34.5 gene loci [15]. GM-CSF functions as a potent immune stimulator by promoting the recruitment and maturation of DCs and macrophages [15]. It is not clear what contribution the GM-CSF makes to anti-tumor efficacy of T-VEC™ as other vectors have shown efficacy in the absence of cytokine transgenes [9].
T-VEC demonstrated strong lytic activity when tested in vitro in human tumor cell lines, including melanoma and pancreatic cancer cells [15]. In mice, intratumoral injection of A20 lymphoma tumors with T-VEC resulted in significant reduction of tumor volume of both injected and non-injected tumors, and treated mice rejected subsequent tumor re-challenge [15]. Corroborating the aforementioned study, a recent study in mice found similar systemic immune responses induced by T-VEC to distal untreated tumors [21]. Furthermore, a significant increase of CD3+ and CD8+ T cells was observed in both injected and contralateral tumors and these T cells were tumor specific [21]. In humans, TVEC monotherapy has also demonstrated distant responses in untreated tumors [22]. Intralesional injection of T-VEC altered the TME and increased MART-1 (melanoma antigen recognized by T-cells 1) specific CD8+ T-cell infiltration in melanoma patients [22]. There was also evidence of these cytotoxic T cells in the peripheral blood of treated patients, suggesting the induction of systemic anti-tumor immunity [23][22]. In addition, injected lesions demonstrated a decrease in Tregs, T-suppressor cells, and MDSCs [22]. In another study, T-VEC in combination with anti-PD-1 antibody, pembrolizumab, altered the TME and increased cytotoxic CD8+ T cell infiltration [23]. These findings demonstrate that OVT can improve the efficacy of ICIs as a combination therapy.
The G207 strain is an attenuated HSV-1 OV based on the HSV-1 (F) strain, which contains deletions of both copies of the ICP34.5 gene, as well as an Escherichia coli lacZ gene insertion which inactivates the ICP6 gene [24]. The ICP6 gene encodes for a subunit of ribonucleotide reductase, an enzyme necessary for viral DNA synthesis in quiescent cells [25]. The inactivation of the ICP6 gene restricts the virus to replicate in actively dividing cells, thus giving the virus selectivity for tumor cells [24].
Early reports demonstrated the utility of G207 as an in situ cancer vaccine [26]. In this study, G207, after intratumoral inoculation of colorectal carcinoma cells, was reported to induce an anti-tumor immune response. Further, in mice that had two tumors engrafted, intratumoral inoculation with G207 reduced tumor growth rates in treated and contralaterally engrafted but untreated tumors, demonstrating an abscopal effect [26]. Intratumoral inoculation with G207 in pre-clinical models of U-87MG gliomas resulted in a significant reduction of tumor volume and prolonged survival [24]. Tumor vasculatures were destroyed in mice bearing rhabdomyosarcoma xenografts following intratumoral treatment with G207 [27]. In the same line, work by Huszthy et al. in athymic nude rats implanted with human glioblastoma (GBM) biopsies further suggests that G207 may possess antiangiogenic properties [28]. The authors also observed accumulated CD68+ microglia cells and macrophages within the tumor bed around viral plaques in G207 injected tumors [28]. In this study, viral plaques and CD68+ cells within the tumor tissue were confirmed by immunostaining [28].
In Phase 1b clinical trials with patients suffering from recurrent GBM, G207 was introduced directly into resected cavities after tumors were removed surgically [29]. G207 demonstrated high safety profiles with post treatment evidence of significant infiltration of CD8+ T cells and macrophages/microglia into the TME [29]. Most recently, results from the first in-human trial of G207 for malignant pediatric cerebellar brain tumors were reported [30][31]. Patients had catheters introduced which allowed direct administration of virus to tumors. Results were highly encouraging with only a single non-responder of 12 patients enrolled. There was substantial evidence of lymphocyte infiltration into tumors in some patients [31]. Interestingly, when patients who were HSV-1 seropositive were excluded from analysis, increased circulating numbers of natural killer (NK) cells was found to positively correlate with survival. Median survival for front-line therapies of high-grade pediatric gliomas is 5.6 months [32][33]. While not directly comparable, in this trial, G207 treatment resulted in median survival of 12.2 months [7]. Moreover, at the time of reporting four of the patients were still surviving 18 months after treatment with G207. This result exceeded expectations and provides strong support for Phase 2 trials.
HSV1716 is derived from HSV-1 strain 17 and lacks both copies of the ICP34.5 genes [34]. HSV1716 OVT has been demonstrated to reduce tumor growth and provide survival advantage as well as to induce significant tumor infiltration of inflammatory immune cells, including CD4+ T cells, CD8+ T cells, NK cells and macrophages in mouse models of intracranial M-3 S91 Cloudman melanoma, 4T1 breast, and rhabdomyosarcoma cancers [35][36][37]. One mechanism used by HSV1716 to recruit effector immune cells into the TME is through the upregulation of chemokines: IFN-γ-inducible protein-10 (IP-10 also known as CXCL10) and monokine induced by IFN-γ (MIG also known as CXCL9) [38]. IP-10 and MIG are known to have chemotactic effects on activated T cells and NK cells. Recent studies in PyMT-TS1 breast cancer model further demonstrated HSV1716’s ability to alter the TME immunosuppressive milieu. In this study, HSV1716 OVT resulted to reduced number of Tregs and reprogrammed M2-like TAMs to a more pro-inflammatory M1-like phenotype in the TME when compared to matched controls [39].
HSV1716 has been evaluated in a number of clinical trials as an intervention strategy for melanoma, mesothelioma, non-CNS solid tumors, and glioma [40][41][42][43]. For mesothelioma, patients were administered HSV1716 via an intrapleural catheter [43]. In the majority of patients after treatment with HSV1716, levels of Th1 cytokines IL-2, IFN-g, TNFa in pleural fluid were increased 5–10-fold [43]. Interestingly, the authors reported the development of novel anti-tumoral antibody responses in four of the patients treated with HSV1716 [43]. In a previous study, HSV1716 was administered intravenously in young patients with extra-cranial solid tumors [41]. Impressively, intravenous administration at up to 107 infectious doses was well tolerated and two patients exhibited stable disease. Unfortunately, TMEs were not studied in biopsied specimens. In another study in young patients with brain tumors, up to 107 infectious units were administered intratumorally [41]. Administration was well tolerated. No biopsies were performed, but imaging revealed metabolic activity consistent with inflammatory responses.
HF10 is a non-engineered, spontaneously mutated OV derived from the HSV-1 HF strain. At the genome level, HF10 lacks the expression of functional unique long (UL) 43, UL49.5, UL55, UL56, and latency-associated transcripts (LAT), and overexpresses UL53 and UL54 genes [44]. The functions of these altered genes and how they may contribute to the antitumor efficacy of HF10 have been reviewed [44]. In a C3H mouse model of head and neck squamous cell carcinoma (SCC VII), intratumoral injection of HF10 reduced tumor growth and significantly enhanced survival [45]. Immunohistochemistry analysis showed evidence of tumor necrosis and infiltration of CD8+ T cells around HSV-infected cells in HF10 treated tumors [45]. These findings correspond with the results of previous studies of HF10 OVT against other malignancies [46], however, the authors did not show whether HF10 as a monotherapy, altered the immunosuppressive cell populations in the TME.
There have been several clinical trials to evaluate the safety and efficacy of HF10 for a number of different cancers including head and neck squamous cell carcinoma (HNSCC), breast cancer, melanoma and pancreatic cancer [44][47][48][49][50][51]. All trials report that HF10 was safe and tolerable in each trial. Analysis of infiltrating immune cells was reported in a subset of these trials. In a recent Phase I trial of nonresectable pancreatic cancer where patients were treated intratumorally with HF10, immunohistochemistry on HF10 treated tumors revealed a statistically significant infiltration of CD8+ T-cells compared to tumors that did not receive HF10 [51]. Analysis of CD4+ T-cells revealed no statistically significant differences between treated and untreated tumors. The study also included analysis of macrophages and other APCs. Of these, macrophages were found to infiltrate tumors in statistically significant numbers in HF10 treated tumors relative to untreated tumors. Consistent with a trial for G207 [31], the authors noted an increase in circulating NK cells in patients treated with HF10.
NV1020 is derived from the HSV-1 strain F designated as R7020, that was originally developed as a vaccine against HSV-1 and HSV-2 infection [52]. The NV1020 genome contains a 15-kb deletion region at the UL/S junction, which encompasses genes encoding ICP0, ICP4, and ICP34.5, as well as UL56 [53]. NV1020 is further attenuated by a 700-bp deletion in the tk gene locus that prevents the expression of the overlapping UL24 gene [53]. In addition, the virus carries an insertion of an exogenous copy of the HSV-1 tk gene, and a 5.2-kb fragment of HSV-2 DNA [53]. NV1020 replicates efficiently in transformed cells and the insertion of the tk gene ensures that viral infection or toxicity can be controlled with antiviral drugs, like acyclovir.
The safety and efficacy profile of NV1020 in the treatment of peritoneally disseminated gastric cancer has previously been investigated in preclinical models. When tumor bearing mice were treated intraperitoneally with NV1020, it significantly reduced tumor burdens and conferred significant survival advantage when compared to control treated animals [54]. In addition, when the brain, liver, kidneys and tumor of NV1020 treated mice were evaluated for HSV biodistribution and necrosis, immunohistochemistry analysis showed no virus or necrosis in any non-tumor tissues [54]. However, significant HSV staining and extensive necrosis was observed in tumor specimens. Furthermore, NV1020 has also been shown to have significant promise in the treatment of other pre-clinical cancer models, including pancreatic, pleural, and bladder cancer, as well as head and neck squamous cell carcinoma [55][56][57][58].
The safety and tolerability of NV1020 was first evaluated in a Phase 1 study involving patients with colorectal cancer metastatic to liver (mCRC) [59]. NV1020 was administered to 12 patients (3 per cohort) via intrahepatic arterial infusion in escalating dose of 3 × 106, 1 × 107, 3 × 107, and 1 × 108 PFU. All patients received cycles of floxuridine through a chemotherapy infusion pump with several combinations of chemotherapy (irinotecan, 5-fluorouracil, leucovorin, or oxaliplatin) 1 and 2 months after administration of virus. Most frequent adverse events associated with the administration of NV1020 included pyrexia, headache and rigor. Although, one patient had a severe case of increased gamma glutamyl transferase (GGT) levels occurring 12 h after virus infusion. No significant change in serum levels of cytokines and T cells due NV1020 administration was observed. Radiologic assessment of anti-tumor activity 28 days after viral administration showed that two patients had reduction in tumor size of 39% (1 × 108 PFU cohort) and 20% (3 × 107 PFU cohort), respectively. In addition, 3 patients demonstrated progression of disease, while 7 patients had disease stabilization.
A subsequent study aimed to evaluate the therapeutic effects of an optimally tolerated dose of NV1020 in patients with mCRC has been completed. According to the results, treatment with virus through hepatic artery infusion was well tolerated and was associated with stable disease in 50% of the patients. Immunologically, individual infusions of NV1020 induced a dose-related increase in the levels of IL-6, TNF-α, and IFN-γ. The addition of chemotherapy resulted to a median time to progression (TTP) and a median overall survival (OS) of 6.4 months and 11.8 months respectively. Altogether, this study suggests that NV1020 can stabilize liver metastases and sensitize tumors to chemotherapy resulting prolonged overall survival in patients with mCRC [60].
2.2. Interactions of oHSV with the Tumor Vasculature
Tumor vasculatures restrain the migration of immune cells from reaching tumor targets through the expression of immunosuppressive ligands and the downregulation of adhesion molecules [61]. Studies on how oHSV-derived OVT disrupts tumor-induced vasculature has demonstrated mixed results. On one hand, studies have shown a direct anti-angiogenic effects of oHSV [62]. Although most of the reported anti-angiogenic oHSV vectors are armed with angiostatic factors or angiogenesis inhibitors. One example is the oHSV RAMBO (rapid antiangiogenesis mediated by oncolytic virus). Hardcastle et al. hypothesized that incorporating an extracellular fragment of the brain-specific angiogenesis inhibitor 1 (BAI1), vasculostatin (Vstat120), in RAMBO would counter the downregulation of thrombospondin 1 (TSP-1) and the increased cysteine-rich 61 (CYR61) integrin activation in the TME, thereby enhancing the anti-tumor efficacy [63]. Indeed, RAMBO treatment promoted significant anti-tumor and anti-angiogenic responses over the control virus, in the treatment of mice bearing intracranial and subcutaneous gliomas. In addition, RAMBO-treated tumors showed a significant reduction in tumor microvessel density (MVD) and vascular volume fraction (VVF) [63]. Similarly, Tsuji et al. demonstrated that treatment with T-TSP-1, an oHSV armed with human TSP-1, reduced tumor MVD and exerted an enhanced anti-tumor efficacy against human gastric cancer in vivo [64].
On the other hand, other reports have suggested that oHSVs induce pro-angiogenic responses [65][66]. The downregulation of TSP-1, an anti-angiogenic factor, and the increased expression of CYR61, a pro-angiogenic factor, in infected cells are some of the limitations of the neovascular responses associated with oHSV infection [65][66]. When taken together, there is no clear census regarding the anti-angiogenic or pro-angiogenic nature of oHSV vectors.
2.3. Interactions of oHSV with the Extracellular Matrix of Tumors
The extracellular matrix (ECM) of tumors controls a number of important regulatory processes central to tumor pathology [67]. However, its function as a barrier to prevent viral replication and spread has motivated a number of groups to manipulate the ECM as a component of OVT [68][69][70][71][72][73]. These efforts include the exogenous addition of enzymes to degrade the ECM during OVT and the engineering of novel viruses that express matrix degrading enzymes. The molecular makeup of the ECM can be unique to each tumor. As such, the approaches to disrupt the ECM as a component of OVT may not represent a “one size fits all” solution. Further, ECM remodeling proteins such as matrix metalloproteinases (MMPs), while generally understood to be protumoral, can have anti-tumoral effects depending on individual tumor characteristics [74]. Bearing this in mind, a number of groups have been successful incorporating ECM-targeting approaches for OVT.
Building on earlier work that demonstrated exogenous addition of matrix metalloproteinase 9 (MMP9) in neuroblastoma cells enhanced oHSV spread [68], Sette et al. created a novel oHSV that expresses MMP9 (KMMP9) [75]. MMP9 degrades Type IV collagen which is present in the ECM of glioblastoma multiforme (GBM). KMMP9 possessed enhanced spread in GBM spheroids and treatment of intracranial mouse xenografts with KMMP9 resulted in increased survival compared to xenografts treated with control non-MMP9 expressing-virus [75].
Mckee et al. demonstrated that targeting collagen using bacterial collagenases enhanced efficacy of OVT in melanoma xenograft models [71]. Bacterial collagenases were introduced intratumorally, coincident with MGH2, an oHSV vector. The combined effect was enhanced intratumoral spread of virus and significantly reduced tumor growth rates.
In brain tumors many CSPGs are upregulated and this upregulation is associated with the enhanced pathogenicity of these tumors. Sugar modifications on CSPGs are responsible for limiting diffusion in the ECM. Dmitrievea et al. reasoned that removal of these moieties with a bacterial enzyme, chondroitinase ABC (chase-ABC), would facilitate replication and spread of oHSV through CNS tumors [70]. To this end, they engineered an oHSV (OV-Chase) to express Chase-ABC. OV-Chase significantly enhanced replication and spread through subcutaneous gliomas, reduced tumor growth rates and increased survival in mouse glioma xenografts.
More direct approaches have been taken to subvert the ECM barrier to viral replication and spread through tumors. E-cadherin is a molecule that facilitates cellular adhesion through nectin-1. Nectin-1 is the predominant receptor for HSV-1 infection and the addition of E-cadherin was hypothesized to directly increase viral infection and spread by promoting receptor binding [69]. This group engineered an oHSV (OVCDH1) to express human E-cadherin. OVCDH1 exhibited enhanced spread through GBM cells in vitro, reduced tumor growth rates in vivo and enhanced survival in xenograft and immunocompetent models of GBM.
Most studies of the effect of manipulating the ECM on OVT efficacy have focused on viral replication and spread. There has been relatively little effort spent to look at the effect manipulating the ECM during OVT has on immune cell infiltration. The above study is particularly interesting in this respect, as they examined the effect of E-cadherin on infiltrating NK cells, macrophage, microglia and T-cells. Perhaps due to an increase in cytolytic activity of OVCDH1 this group noted an increase in NK cell infiltration into tumors after OVCDH1 treatment versus control virus treatment. No other differences in immune cell infiltration were observed between OVCDH1 and control groups.
Finally, while the above groups demonstrated that overexpression of ECM proteins can benefit OVT efficacy, Mahller et al., reasoned that inhibition of ECM protein activity may benefit OVT [72]. This group hypothesized that, as the function of many ECM protein activities are pro-tumoral, using a virus to express TIMP-3, a matrix metalloprotein inhibitor, might benefit oHSV-derived OVT. TIMP-3, tissue inhibitor of MMPs 3, is an inhibitor of all MMPs. Treatment with TIMP-3 reduced tumor growth rates in a number of cancer models [76][77][78]. Using this rationale, Mahller et al., engineered the G207 oHSV (described below and in Table 1) to express TIMP-3 and called it rQT3. Compared to control virus which expresses luciferase, treatment of neuroblastoma and malignant peripheral nerve sheath (MPNST) xenografts with rQT3 was superior at reducing tumor growth rates and enhancing survival.
Table 1. Summary of HSV-derived Oncolytic Viruses Discussed.
| Oncolytic Herpes Simplex Virus | Strain | HSV Gene Mutations | Transgene | Reference |
|---|---|---|---|---|
| KMMP9 | KOS | Deletion of residues 2–24 and amino acid substitution, Y38C, in gD. gB:NT |
Human EGFR, miR-124 target in ICP4 3′UTR, MMP9 | [75] |
| rQT3 | F | ICP34.5Δ, ICP6Δ | Human TIMP3 | [79] |
| MGH2 | F | ICP34.5Δ, ICP6Δ | CYP2B1 and human shiCE |
[80][81] |
| OVCDH1 | Q1 | ICP34.5Δ, ICP6Δ | Human CDH1 | [69] |
| OV-Chase | F | ICP34.5Δ, ICP6Δ | Bacterial Choindroitinase ABC | [70] |
| T-VEC | JS-1 | ICP34.5Δ, ICP47Δ | Human GM-CSF | [15] |
| G207 | F | ICP34.5Δ | Escherichia coli lacZ gene at ICP6 | [24] |
| HSV1716 | 17 | ICP34.5Δ | None | [34] |
| HF10 | HF | Reduced expression of UL43, UL49.5, UL55, UL56 and LAT genes; increased expression of UL53, and UL54 genes | None | [44] |
| NV1020 | F | Joint deletion (1 copy of ICP0, ICP4, ICP 34.5), UL56Δ, UL5/6 duplication, UL24Δ, TKΔ |
TK under ICP4 promoter control, 5.2-kb fragment of HSV-2 DNA |
[53] |
| G47Δ-mIL-12 | F | ICP34.5Δ, ICP6Δ, ICP47Δ | Mouse IL-12 | [82] |
| VC2 | F | gKΔ, UL20Δ | None | [9] |
| RAMBO | F | ICP34.5Δ, ICP6Δ | Human Vstat120 | [63] |
| T-TSP1 | F | ICP34.5Δ, ICP6Δ, ICP47Δ | Human TSP-1 | [64] |
References
- van der Woude, L.L.; Gorris, M.A.J.; Halilovic, A.; Figdor, C.G.; de Vries, I.J.M. Migrating into the Tumor: A Roadmap for T Cells. Trends Cancer 2017, 3, 797–808.
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964.
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571.
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218.
- Newman, J.H.; Chesson, C.B.; Herzog, N.L.; Bommareddy, P.K.; Aspromonte, S.M.; Pepe, R.; Estupinian, R.; Aboelatta, M.M.; Buddhadev, S.; Tarabichi, S.; et al. Intratumoral injection of the seasonal flu shot converts immunologically cold tumors to hot and serves as an immunotherapy for cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 1119–1128.
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618.
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10.
- Ma, W.; He, H.; Wang, H. Oncolytic herpes simplex virus and immunotherapy. BMC Immunol. 2018, 19, 40.
- Uche, I.K.; Fowlkes, N.; Vu, L.; Watanabe, T.; Carossino, M.; Nabi, R.; Del Piero, F.; Rudd, J.S.; Kousoulas, K.G.; Rider, P.J.F. Novel Oncolytic Herpes Simplex Virus 1 VC2 Promotes Long-Lasting, Systemic Anti-melanoma Tumor Immune Responses and Increased Survival in an Immunocompetent B16F10-Derived Mouse Melanoma Model. J. Virol. 2021, 95.
- Wirsching, H.G.; Zhang, H.; Szulzewsky, F.; Arora, S.; Grandi, P.; Cimino, P.J.; Amankulor, N.; Campbell, J.S.; McFerrin, L.; Pattwell, S.S.; et al. Arming oHSV with ULBP3 drives abscopal immunity in lymphocyte-depleted glioblastoma. JCI Insight 2019, 4.
- Alessandrini, F.; Menotti, L.; Avitabile, E.; Appolloni, I.; Ceresa, D.; Marubbi, D.; Campadelli-Fiume, G.; Malatesta, P. Eradication of glioblastoma by immuno-virotherapy with a retargeted oncolytic HSV in a preclinical model. Oncogene 2019, 38, 4467–4479.
- Russell, L.; Swanner, J.; Jaime-Ramirez, A.C.; Wang, Y.; Sprague, A.; Banasavadi-Siddegowda, Y.; Yoo, J.Y.; Sizemore, G.M.; Kladney, R.; Zhang, J.; et al. PTEN expression by an oncolytic herpesvirus directs T-cell mediated tumor clearance. Nat. Commun. 2018, 9, 5006.
- Leoni, V.; Vannini, A.; Gatta, V.; Rambaldi, J.; Sanapo, M.; Barboni, C.; Zaghini, A.; Nanni, P.; Lollini, P.L.; Casiraghi, C.; et al. A fully-virulent retargeted oncolytic HSV armed with IL-12 elicits local immunity and vaccine therapy towards distant tumors. PLoS Pathog. 2018, 14, e1007209.
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788.
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303.
- Brown, S.M.; MacLean, A.R.; Aitken, J.D.; Harland, J. ICP34.5 influences herpes simplex virus type 1 maturation and egress from infected cells in vitro. J. Gen. Virol. 1994, 75 Pt 12, 3679–3686.
- Chou, J.; Kern, E.R.; Whitley, R.J.; Roizman, B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 1990, 250, 1262–1266.
- He, B.; Gross, M.; Roizman, B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 843–848.
- Goldsmith, K.; Chen, W.; Johnson, D.C.; Hendricks, R.L. Infected cell protein (ICP)47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J. Exp. Med. 1998, 187, 341–348.
- Mulvey, M.; Poppers, J.; Ladd, A.; Mohr, I. A herpesvirus ribosome-associated, RNA-binding protein confers a growth advantage upon mutants deficient in a GADD34-related function. J. Virol. 1999, 73, 3375–3385.
- Moesta, A.K.; Cooke, K.; Piasecki, J.; Mitchell, P.; Rottman, J.B.; Fitzgerald, K.; Zhan, J.; Yang, B.; Le, T.; Belmontes, B.; et al. Local Delivery of OncoVEX(mGM-CSF) Generates Systemic Antitumor Immune Responses Enhanced by Cytotoxic T-Lymphocyte-Associated Protein Blockade. Clin. Cancer Res. 2017, 23, 6190–6202.
- Kaufman, H.L.; Kim, D.W.; DeRaffele, G.; Mitcham, J.; Coffin, R.S.; Kim-Schulze, S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann. Surg. Oncol. 2010, 17, 718–730.
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.
- Mineta, T.; Rabkin, S.D.; Yazaki, T.; Hunter, W.D.; Martuza, R.L. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1995, 1, 938–943.
- Goldstein, D.J.; Weller, S.K. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: Isolation and characterization of an ICP6 lacZ insertion mutant. J. Virol. 1988, 62, 196–205.
- Toda, M.; Rabkin, S.D.; Kojima, H.; Martuza, R.L. Herpes simplex virus as an in situ cancer vaccine for the induction of specific anti-tumor immunity. Hum. Gene Ther. 1999, 10, 385–393.
- Cinatl, J., Jr.; Michaelis, M.; Driever, P.H.; Cinatl, J.; Hrabeta, J.; Suhan, T.; Doerr, H.W.; Vogel, J.U. Multimutated herpes simplex virus g207 is a potent inhibitor of angiogenesis. Neoplasia 2004, 6, 725–735.
- Huszthy, P.C.; Immervoll, H.; Wang, J.; Goplen, D.; Miletic, H.; Eide, G.E.; Bjerkvig, R. Cellular effects of oncolytic viral therapy on the glioblastoma microenvironment. Gene Ther. 2010, 17, 202–216.
- Markert, J.M.; Liechty, P.G.; Wang, W.; Gaston, S.; Braz, E.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Lakeman, A.D.; Palmer, C.A.; et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol. Ther. 2009, 17, 199–207.
- Bernstock, J.D.; Bag, A.K.; Fiveash, J.; Kachurak, K.; Elsayed, G.; Chagoya, G.; Gessler, F.; Valdes, P.A.; Madan-Swain, A.; Whitley, R.; et al. Design and Rationale for First-in-Human Phase 1 Immunovirotherapy Clinical Trial of Oncolytic HSV G207 to Treat Malignant Pediatric Cerebellar Brain Tumors. Hum. Gene Ther. 2020, 31, 1132–1139.
- Friedman, G.K.; Johnston, J.M.; Bag, A.K.; Bernstock, J.D.; Li, R.; Aban, I.; Kachurak, K.; Nan, L.; Kang, K.D.; Totsch, S.; et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N. Engl. J. Med. 2021.
- Jakacki, R.I.; Cohen, K.J.; Buxton, A.; Krailo, M.D.; Burger, P.C.; Rosenblum, M.K.; Brat, D.J.; Hamilton, R.L.; Eckel, S.P.; Zhou, T.; et al. Phase 2 study of concurrent radiotherapy and temozolomide followed by temozolomide and lomustine in the treatment of children with high-grade glioma: A report of the Children’s Oncology Group ACNS0423 study. Neuro Oncol. 2016, 18, 1442–1450.
- Kline, C.; Felton, E.; Allen, I.E.; Tahir, P.; Mueller, S. Survival outcomes in pediatric recurrent high-grade glioma: Results of a 20-year systematic review and meta-analysis. J. Neurooncol. 2018, 137, 103–110.
- Dolan, A.; McKie, E.; MacLean, A.R.; McGeoch, D.J. Status of the ICP34.5 gene in herpes simplex virus type 1 strain 17. J. Gen. Virol. 1992, 73 Pt 4, 971–973.
- Miller, C.G.; Fraser, N.W. Role of the immune response during neuro-attenuated herpes simplex virus-mediated tumor destruction in a murine intracranial melanoma model. Cancer Res. 2000, 60, 5714–5722.
- Thomas, D.L.; Fraser, N.W. HSV-1 therapy of primary tumors reduces the number of metastases in an immune-competent model of metastatic breast cancer. Mol. Ther. 2003, 8, 543–551.
- Chen, C.Y.; Wang, P.Y.; Hutzen, B.; Sprague, L.; Swain, H.M.; Love, J.K.; Stanek, J.R.; Boon, L.; Conner, J.; Cripe, T.P. Cooperation of Oncolytic Herpes Virotherapy and PD-1 Blockade in Murine Rhabdomyosarcoma Models. Sci. Rep. 2017, 7, 2396.
- Benencia, F.; Courreges, M.C.; Conejo-Garcia, J.R.; Mohamed-Hadley, A.; Zhang, L.; Buckanovich, R.J.; Carroll, R.; Fraser, N.; Coukos, G. HSV oncolytic therapy upregulates interferon-inducible chemokines and recruits immune effector cells in ovarian cancer. Mol. Ther. 2005, 12, 789–802.
- Kwan, A.; Winder, N.; Atkinson, E.; Al-Janabi, H.; Allen, R.J.; Hughes, R.; Moamin, M.; Louie, R.; Evans, D.; Hutchinson, M.; et al. Macrophages Mediate the Antitumor Effects of the Oncolytic Virus HSV1716 in Mammary Tumors. Mol. Cancer Ther. 2020.
- MacKie, R.M.; Stewart, B.; Brown, S.M. Intralesional injection of herpes simplex virus 1716 in metastatic melanoma. Lancet 2001, 357, 525–526.
- Streby, K.A.; Geller, J.I.; Currier, M.A.; Warren, P.S.; Racadio, J.M.; Towbin, A.J.; Vaughan, M.R.; Triplet, M.; Ott-Napier, K.; Dishman, D.J.; et al. Intratumoral Injection of HSV1716, an Oncolytic Herpes Virus, Is Safe and Shows Evidence of Immune Response and Viral Replication in Young Cancer Patients. Clin. Cancer Res. 2017, 23, 3566–3574.
- Streby, K.A.; Currier, M.A.; Triplet, M.; Ott, K.; Dishman, D.J.; Vaughan, M.R.; Ranalli, M.A.; Setty, B.; Skeens, M.A.; Whiteside, S.; et al. First-in-Human Intravenous Seprehvir in Young Cancer Patients: A Phase 1 Clinical Trial. Mol. Ther. 2019, 27, 1930–1938.
- Danson, S.J.; Conner, J.; Edwards, J.G.; Blyth, K.G.; Fisher, P.M.; Muthana, M.; Salawu, A.; Taylor, F.; Hodgkinson, E.; Joyce, P.; et al. Oncolytic herpesvirus therapy for mesothelioma—A phase I/IIa trial of intrapleural administration of HSV1716. Lung Cancer 2020, 150, 145–151.
- Eissa, I.R.; Naoe, Y.; Bustos-Villalobos, I.; Ichinose, T.; Tanaka, M.; Zhiwen, W.; Mukoyama, N.; Morimoto, T.; Miyajima, N.; Hitoki, H.; et al. Genomic Signature of the Natural Oncolytic Herpes Simplex Virus HF10 and Its Therapeutic Role in Preclinical and Clinical Trials. Front. Oncol. 2017, 7, 149.
- Esaki, S.; Goshima, F.; Ozaki, H.; Takano, G.; Hatano, Y.; Kawakita, D.; Ijichi, K.; Watanabe, T.; Sato, Y.; Murata, T.; et al. Oncolytic activity of HF10 in head and neck squamous cell carcinomas. Cancer Gene Ther. 2020, 27, 585–598.
- Watanabe, D.; Goshima, F.; Mori, I.; Tamada, Y.; Matsumoto, Y.; Nishiyama, Y. Oncolytic virotherapy for malignant melanoma with herpes simplex virus type 1 mutant HF10. J. Dermatol. Sci. 2008, 50, 185–196.
- Hirooka, Y.; Kasuya, H.; Ishikawa, T.; Kawashima, H.; Ohno, E.; Villalobos, I.B.; Naoe, Y.; Ichinose, T.; Koyama, N.; Tanaka, M.; et al. A Phase I clinical trial of EUS-guided intratumoral injection of the oncolytic virus, HF10 for unresectable locally advanced pancreatic cancer. BMC Cancer 2018, 18, 596.
- Nakao, A.; Kimata, H.; Imai, T.; Kikumori, T.; Teshigahara, O.; Nagasaka, T.; Goshima, F.; Nishiyama, Y. Intratumoral injection of herpes simplex virus HF10 in recurrent breast cancer. Ann. Oncol. 2004, 15, 988–989.
- Kimata, H.; Imai, T.; Kikumori, T.; Teshigahara, O.; Nagasaka, T.; Goshima, F.; Nishiyama, Y.; Nakao, A. Pilot study of oncolytic viral therapy using mutant herpes simplex virus (HF10) against recurrent metastatic breast cancer. Ann. Surg. Oncol. 2006, 13, 1078–1084.
- Fujimoto, Y.; Mizuno, T.; Sugiura, S.; Goshima, F.; Kohno, S.; Nakashima, T.; Nishiyama, Y. Intratumoral injection of herpes simplex virus HF10 in recurrent head and neck squamous cell carcinoma. Acta Otolaryngol. 2006, 126, 1115–1117.
- Nakao, A.; Kasuya, H.; Sahin, T.T.; Nomura, N.; Kanzaki, A.; Misawa, M.; Shirota, T.; Yamada, S.; Fujii, T.; Sugimoto, H.; et al. A phase I dose-escalation clinical trial of intraoperative direct intratumoral injection of HF10 oncolytic virus in non-resectable patients with advanced pancreatic cancer. Cancer Gene Ther. 2011, 18, 167–175.
- Meignier, B.; Longnecker, R.; Roizman, B. In vivo behavior of genetically engineered herpes simplex viruses R7017 and R7020: Construction and evaluation in rodents. J. Infect. Dis. 1988, 158, 602–614.
- Kelly, K.J.; Wong, J.; Fong, Y. Herpes simplex virus NV1020 as a novel and promising therapy for hepatic malignancy. Expert Opin. Investig. Drugs 2008, 17, 1105–1113.
- Bennett, J.J.; Delman, K.A.; Burt, B.M.; Mariotti, A.; Malhotra, S.; Zager, J.; Petrowsky, H.; Mastorides, S.; Federoff, H.; Fong, Y. Comparison of safety, delivery, and efficacy of two oncolytic herpes viruses (G207 and NV1020) for peritoneal cancer. Cancer Gene Ther. 2002, 9, 935–945.
- McAuliffe, P.F.; Jarnagin, W.R.; Johnson, P.; Delman, K.A.; Federoff, H.; Fong, Y. Effective treatment of pancreatic tumors with two multimutated herpes simplex oncolytic viruses. J. Gastrointest Surg. 2000, 4, 580–588.
- Ebright, M.I.; Zager, J.S.; Malhotra, S.; Delman, K.A.; Weigel, T.L.; Rusch, V.W.; Fong, Y. Replication-competent herpes virus NV1020 as direct treatment of pleural cancer in a rat model. J. Thorac. Cardiovasc. Surg. 2002, 124, 123–129.
- Cozzi, P.J.; Malhotra, S.; McAuliffe, P.; Kooby, D.A.; Federoff, H.J.; Huryk, B.; Johnson, P.; Scardino, P.T.; Heston, W.D.; Fong, Y. Intravesical oncolytic viral therapy using attenuated, replication-competent herpes simplex viruses G207 and Nv1020 is effective in the treatment of bladder cancer in an orthotopic syngeneic model. FASEB J. 2001, 15, 1306–1308.
- Wong, R.J.; Kim, S.H.; Joe, J.K.; Shah, J.P.; Johnson, P.A.; Fong, Y. Effective treatment of head and neck squamous cell carcinoma by an oncolytic herpes simplex virus. J. Am. Coll. Surg. 2001, 193, 12–21.
- Kemeny, N.; Brown, K.; Covey, A.; Kim, T.; Bhargava, A.; Brody, L.; Guilfoyle, B.; Haag, N.P.; Karrasch, M.; Glasschroeder, B.; et al. Phase I, open-label, dose-escalating study of a genetically engineered herpes simplex virus, NV1020, in subjects with metastatic colorectal carcinoma to the liver. Hum. Gene Ther. 2006, 17, 1214–1224.
- Geevarghese, S.K.; Geller, D.A.; de Haan, H.A.; Horer, M.; Knoll, A.E.; Mescheder, A.; Nemunaitis, J.; Reid, T.R.; Sze, D.Y.; Tanabe, K.K.; et al. Phase I/II study of oncolytic herpes simplex virus NV1020 in patients with extensively pretreated refractory colorectal cancer metastatic to the liver. Hum. Gene Ther. 2010, 21, 1119–1128.
- Motz, G.T.; Coukos, G. Deciphering and reversing tumor immune suppression. Immunity 2013, 39, 61–73.
- Benencia, F.; Courreges, M.C.; Conejo-Garcia, J.R.; Buckanovich, R.J.; Zhang, L.; Carroll, R.H.; Morgan, M.A.; Coukos, G. Oncolytic HSV exerts direct antiangiogenic activity in ovarian carcinoma. Hum. Gene Ther. 2005, 16, 765–778.
- Hardcastle, J.; Kurozumi, K.; Dmitrieva, N.; Sayers, M.P.; Ahmad, S.; Waterman, P.; Weissleder, R.; Chiocca, E.A.; Kaur, B. Enhanced antitumor efficacy of vasculostatin (Vstat120) expressing oncolytic HSV-1. Mol. Ther. 2010, 18, 285–294.
- Tsuji, T.; Nakamori, M.; Iwahashi, M.; Nakamura, M.; Ojima, T.; Iida, T.; Katsuda, M.; Hayata, K.; Ino, Y.; Todo, T.; et al. An armed oncolytic herpes simplex virus expressing thrombospondin-1 has an enhanced in vivo antitumor effect against human gastric cancer. Int. J. Cancer 2013, 132, 485–494.
- Kurozumi, K.; Hardcastle, J.; Thakur, R.; Shroll, J.; Nowicki, M.; Otsuki, A.; Chiocca, E.A.; Kaur, B. Oncolytic HSV-1 infection of tumors induces angiogenesis and upregulates CYR61. Mol. Ther. 2008, 16, 1382–1391.
- Aghi, M.; Rabkin, S.D.; Martuza, R.L. Angiogenic response caused by oncolytic herpes simplex virus-induced reduced thrombospondin expression can be prevented by specific viral mutations or by administering a thrombospondin-derived peptide. Cancer Res. 2007, 67, 440–444.
- Henke, E.; Nandigama, R.; Ergun, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2019, 6, 160.
- Balz, K.; Trassl, L.; Hartel, V.; Nelson, P.P.; Skevaki, C. Virus-Induced T Cell-Mediated Heterologous Immunity and Vaccine Development. Front. Immunol. 2020, 11, 513.
- Xu, B.; Ma, R.; Russell, L.; Yoo, J.Y.; Han, J.; Cui, H.; Yi, P.; Zhang, J.; Nakashima, H.; Dai, H.; et al. An oncolytic herpesvirus expressing E-cadherin improves survival in mouse models of glioblastoma. Nat. Biotechnol. 2018.
- Dmitrieva, N.; Yu, L.; Viapiano, M.; Cripe, T.P.; Chiocca, E.A.; Glorioso, J.C.; Kaur, B. Chondroitinase ABC I-mediated enhancement of oncolytic virus spread and antitumor efficacy. Clin. Cancer Res. 2011, 17, 1362–1372.
- McKee, T.D.; Grandi, P.; Mok, W.; Alexandrakis, G.; Insin, N.; Zimmer, J.P.; Bawendi, M.G.; Boucher, Y.; Breakefield, X.O.; Jain, R.K. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006, 66, 2509–2513.
- Mahller, Y.Y.; Vaikunth, S.S.; Ripberger, M.C.; Baird, W.H.; Saeki, Y.; Cancelas, J.A.; Crombleholme, T.M.; Cripe, T.P. Tissue inhibitor of metalloproteinase-3 via oncolytic herpesvirus inhibits tumor growth and vascular progenitors. Cancer Res. 2008, 68, 1170–1179.
- Everts, A.; Bergeman, M.; McFadden, G.; Kemp, V. Simultaneous Tumor and Stroma Targeting by Oncolytic Viruses. Biomedicines 2020, 8, 474.
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27.
- Sette, P.; Amankulor, N.; Li, A.; Marzulli, M.; Leronni, D.; Zhang, M.; Goins, W.F.; Kaur, B.; Bolyard, C.; Cripe, T.P.; et al. GBM-Targeted oHSV Armed with Matrix Metalloproteinase 9 Enhances Anti-tumor Activity and Animal Survival. Mol. Ther. Oncolytics 2019, 15, 214–222.
- Anania, M.C.; Sensi, M.; Radaelli, E.; Miranda, C.; Vizioli, M.G.; Pagliardini, S.; Favini, E.; Cleris, L.; Supino, R.; Formelli, F.; et al. TIMP3 regulates migration, invasion and in vivo tumorigenicity of thyroid tumor cells. Oncogene 2011, 30, 3011–3023.
- Lin, H.; Zhang, Y.; Wang, H.; Xu, D.; Meng, X.; Shao, Y.; Lin, C.; Ye, Y.; Qian, H.; Wang, S. Tissue inhibitor of metalloproteinases-3 transfer suppresses malignant behaviors of colorectal cancer cells. Cancer Gene Ther. 2012, 19, 845–851.
- Chetty, C.; Lakka, S.S.; Bhoopathi, P.; Kunigal, S.; Geiss, R.; Rao, J.S. Tissue inhibitor of metalloproteinase 3 suppresses tumor angiogenesis in matrix metalloproteinase 2-down-regulated lung cancer. Cancer Res. 2008, 68, 4736–4745.
- Terada, K.; Wakimoto, H.; Tyminski, E.; Chiocca, E.A.; Saeki, Y. Development of a rapid method to generate multiple oncolytic HSV vectors and their in vivo evaluation using syngeneic mouse tumor models. Gene Ther. 2006, 13, 705–714.
- Tyminski, E.; Leroy, S.; Terada, K.; Finkelstein, D.M.; Hyatt, J.L.; Danks, M.K.; Potter, P.M.; Saeki, Y.; Chiocca, E.A. Brain tumor oncolysis with replication-conditional herpes simplex virus type 1 expressing the prodrug-activating genes, CYP2B1 and secreted human intestinal carboxylesterase, in combination with cyclophosphamide and irinotecan. Cancer Res. 2005, 65, 6850–6857.
- MacLean, A.R.; ul-Fareed, M.; Robertson, L.; Harland, J.; Brown, S.M. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the ‘a’ sequence. J. Gen. Virol. 1991, 72 Pt 3, 631–639.
- Cheema, T.A.; Wakimoto, H.; Fecci, P.E.; Ning, J.; Kuroda, T.; Jeyaretna, D.S.; Martuza, R.L.; Rabkin, S.D. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc. Natl. Acad. Sci. USA 2013, 110, 12006–12011.
More
Information
Subjects:
Biotechnology & Applied Microbiology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
2 times
(View History)
Update Date:
04 Aug 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No