 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Annalisa Astolfi | + 2290 word(s) | 2290 | 2020-05-08 11:36:46 | | | |
| 2 | Nora Tang | Meta information modification | 2290 | 2020-06-10 16:49:37 | | | | |
| 3 | Nora Tang | -18 word(s) | 2272 | 2020-10-30 12:40:41 | | |
Video Upload Options
Gastrointestinal stromal tumors (GIST) are rare neoplasms of mesenchymal origin arising in the gastrointestinal tract. The vast majority are characterized by mutually-exclusive activating mutations in KIT or Platelet-derived growth factor alpha (PDGFRA) receptors, or less frequently by succinate dehydrogenase complex (SDH) or NF1 inactivation, with very rare cases harboring mutant BRAF or RAS alleles. Approximately 5% of GISTs lack any of such mutations and are called quadruple wild-type (WT) GISTs. Recently, Fibroblast Growth Factor (FGF)/FGF-receptor (FGFR) signaling emerged as an activated pathway in different molecular subgroups of GISTs due to activating mutations, oncogenic gene fusions, or autocrine/paracrine signaling.
1. Introduction
Gastrointestinal stromal tumors (GIST) are the most common mesenchymal tumors of the GI tract, arising from the interstitial cells of Cajal (ICCs) or their precursors. Although the vast majority of GISTs harbors KIT mutations, that confer the highest sensitivity to Tyrosine Kinase Inhibitors (TKIs) or more rarely PDGFRA mutations, about 10–15% of adult cases do not harbor any of these driver mutations and have historically been called as wild-type (WT). Among them, from 20% to 40% show loss of function of the succinate dehydrogenase complex (SDH), therefore defined as SDH-deficient GISTs [1][2][3]. Conversely, about 15% of KIT/PDGFRA WT cases harbor mutations in BRAF/RAS or NF1 and are referred to as RAS-pathway (RAS-P) mutant GISTs [2][4][5][6]. The remaining cases, accounting for about 5% of all GISTs, are usually referred to as KIT/PDGFRA/SDH/RAS-P WT or quadruple WT (qWT) GISTs [7]. Notably, this specific subgroup shares a distinct transcriptome profile that is profoundly different from other GIST subtypes [8]. However, to date a consensus on the recurrent oncogenic driver has still not been found, whereas many different and often private mutational events have been reported, confirming the great molecular heterogeneity of this subgroup of GISTs [9].
Indeed, an ETV6–NTRK3 gene fusion was the first rearrangement to be described [10][11]. Moreover, two fusion genes involving FGFR1 (FGFR1–HOOK3 and FGFR1–TACC1) and other chimeric fusions (KIT–PDGFRA, MARK2–PPFIA1 and SPRED2–NELFCD) have been reported [2][11][12].
The fibroblast growth factor signaling pathway relies on a family of receptor tyrosine kinases (FGFR) and eighteen extracellular ligands (FGFs) and has been implicated in the oncogenic process of different tumor histotypes. It regulates many physiological processes, in both the embryonic and adult stages of development, such as tissue differentiation and homeostasis, angiogenesis, and wound healing [13].
In this scenario, there is a growing interest on FGFR pathway deregulation in GISTs, since FGFR fusion events, together with FGFR mutations and FGFR ligand overexpression represent the most frequent molecular alterations identified in KIT/PDGFRA WT GISTs so far, suggesting its likely pathogenetic role and providing a rationale for targeted therapeutic approaches. Moreover, some evidences on FGFR events and imatinib resistance in KIT/PDGFRA mutant GISTs have been described.
2. FGF/FGFR Family
The human FGFR family consists of four members: FGFR1–4, which are transmembrane receptors with tyrosine kinase activity belonging to the immunoglobulin (Ig) superfamily that can be stimulated and activated by extracellular ligands. FGFR family members display an amino acid sequence highly conserved between members and throughout evolution and differ in their ligand binding capacity and tissue-specific distribution [14]. A fifth family member, lacking the tyrosine kinase domain, FGFR5 or FGFRL1, was discovered on the basis of interaction with FGFR-binding ligands [15]. FGFRL1 is thought to act as a decoy receptor negatively regulating FGFR signaling, therefore inhibiting cellular proliferation and inducing differentiation [15].
Structurally, FGF-receptors are composed of a large extracellular ligand-binding domain, a single transmembrane helix and an intracellular portion containing two split tyrosine kinase domains (Figure 1) [14].
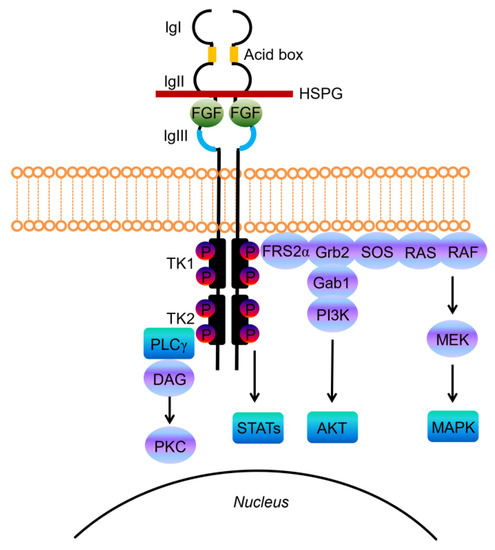
Figure 1. Schematic representations of FGF receptor tyrosine kinase structure, composed of several domains including three extracellular immunoglobulin-like domains (IgI, IgII, and IgIII) and the acid-box, a transmembrane domain, and two intracellular tyrosine kinase domains (TK1 and TK2). The basic structure of the FGF/FGFR complex includes two receptor molecules, two FGFs, and one heparan sulphate proteoglycan (HSPG) chain. The signal transduction network activates four key downstream pathways: RAS/RAF/MAPK, PI3K/AKT, STATs and PLCγ.
The extracellular portion contains three immunoglobulin-like (Ig-like) domains, with a linker region between the first and second Ig-loop containing a highly conserved stretch of glutamate-, aspartate-, and serine-rich sequence, called the acid-box (Figure 1) [16]. The second and third Ig-domains are involved in FGF binding and regulate ligand-binding specificity, while the first one and the acid-box mediate receptor auto-inhibition [14][16].
FGFs, the native ligands of FGF receptors, belong to a family that includes 22 members identified by sequence homology, which were grouped into subfamilies based on function or phylogenetic relation. Eighteen of these ligands encode molecules known to signal through FGF tyrosine kinase receptors. The canonical secreted FGFs can belong to the Fgf1, Fgf4, Fgf7, Fgf8 and Fgf9 subfamily, and exert the prototypical FGF functions that control cell proliferation, differentiation and survival, by binding and activating FGFRs [17]. Canonical FGFs are tightly bound to heparin/heparan sulfate (HS) proteoglycans (HSPGs), which function to limit diffusion through the extracellular matrix (ECM), but also serve as cofactors since cell surface HPSGs stabilizes the FGF ligand–receptor interaction, forming a ternary complex with FGFR that enhances receptor binding and signaling [18].
Upon ligand binding the receptor undergoes dimerization, which leads to a conformational shift in receptor structure that subsequently induces a trans-autophosphorylation event of the intracellular kinase domain, driving the activation of downstream signal transduction pathways (Figure 1) [19].
Deregulation of FGF signaling can be caused by mutations that drive ligand-independent receptor signaling or alterations that support ligand-dependent receptor activation, and is responsible for tumor development and progression or resistance to therapies across many tumor types [13][20][21].
3. FGFR Mutations and Gene Fusions in GIST
Activating alterations in FGF receptors were found only in some cases of the so-called qWT GISTs [17].In fact, in a comprehensive genomic study of a total of 14 qWT GIST patients performed by using whole-exome sequencing (WES), a FGFR1 somatic missense mutation (c.1638C > A; p.N546K) was found in one case, being the only oncogenic event identified [12].
Similarly Shi et al. analyzed 17 KIT/PDGFRA/SDH/RAS-pathway WT GISTs, finding one patient with an activating missense FGFR1 mutation (p.K656E) and two gene fusions: One case harboring FGFR1–HOOK3 (in-frame fusion of FGFR1 intron 17 and HOOK3 intron-4) and two patients carrying FGFR1–TACC1 (in-frame fusion of FGFR1 intron 17 and TACC1 intron-6) [11].
The presence of these clear oncogenic events in tumors lacking any other known molecular alteration strongly supports the driver role of FGFR mutations and gene fusions in a small percentage of qWT GISTs.
4. FGFR Ligand Overexpression
The search of the driver oncogenic event of qWT GISTs led to the very recent identification of the first shared alteration among multiple samples. A high-throughput copy number analysis in eight qWT GISTs highlighted the presence of a recurrent focal gain of one copy in chromosome 11q13.3 cytoband overlapping the FGF3/FGF4 locus in six samples [22]. The cryptic duplication, that was absent in KIT/PDGFRA-mutant and SDH-deficient GISTs, led to the marked overexpression of FGF4 with respect to the other two categories. SDH-deficient GISTs, even if negative for the duplication, showed enhanced FGF4 expression with respect to KIT/PDGFRA-mutant [22]. Given that all quadruple WT GISTs expressed both FGFR1 and FGFR2, and showed phosphorylated-AKT, the existence of an oncogenic autocrine loop supporting tumor growth is conceivable.
Recently, the relevance of FGF-based autocrine loop emerged also in SDH-deficient GISTs. Flavahan et al. in fact showed that genomic hypermethylation induced by SDH loss disrupts CTCF binding at the DNA regions located on the boundaries of the FGF3/FGF4 locus. CTCF is an insulator protein able to bind the boundaries of topologically associated domains (TADs) in the human genome, that are regions where DNA sequences preferentially contact each other and whose genes are generally co-regulated. Hypermethylation prevents CTCF binding, thus disrupting the TAD and allowing the active ANO1 super-enhancer to contact and activate the FGF genes [23]. Therefore, the FGF3–FGF4 locus topology is profoundly altered in SDH-deficient GISTs, with CTCF insulator loss allowing aberrant expression of FGFR ligand genes. As a matter of fact, SDH-deficient GISTs show marked upregulation of FGF3 and FGF4, coupled to high FGFR1 expression and evidence of MAPK signature activation. Moreover, BGJ-398, a potent and selective inhibitor of FGFR1–4, completely suppressed tumor growth in a PDX model derived from an SDH-deficient GIST [23].
These data highlight the relevance of FGF signaling in GISTs devoid of KIT/PDGFRA mutations or RAS-pathway activation, suggesting that FGF4 autocrine loop or FGFR mutations in SDH-deficient and qWT GISTs act as a surrogate of KIT/PDGFRA alterations in sustaining GIST tumor growth (Figure 2).
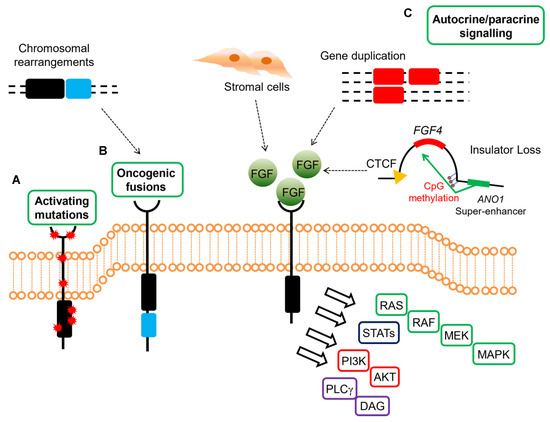
Figure 2. Mechanisms of oncogenic FGFR signaling in gastrointestinal stromal tumors (GISTs). FGFR signaling contributes to oncogenesis through several ligand-dependent and -independent mechanisms in GISTs, signaling through RAS/MAPK, PI3K/AKT, signal transducers and activator of transcription (STATs), phospholipase Cγ (PLCγ). (A) Activating mutations resulting in increased kinase activity in absence of ligand are present in quadruple WT GISTs. (B) Chromosomal translocations drive the fusion of parts of FGFR1 to other genes, thereby resulting in receptor hyperactivation in a ligand-independent manner in quadruple WT GISTs (C) FGFRs can be stimulated by their ligands either in a paracrine fashion, through FGFs produced by stromal cells, or in an autocrine manner, when FGFs are produced directly from the tumor cells that also express FGFRs. In quadruple WT GISTs, FGF4 is overexpressed through genomic duplication of the FGF4 locus, while SDH-deficient GISTs overexpress FGF3/FGF4 by an epigenetic mechanism linked to hyper-methylation of the FGF4 insulator that disrupts CTCF binding, thus allowing the ANO1 super-enhancer to activate the FGF genes. In KIT-mutant GISTs, a role for autocrine or paracrine FGFR signaling has been proposed driving imatinib resistance.
5. FGFR Events and Imatinib Resistance in KIT/PDGFRA Mutant GIST
Although imatinib has revolutionized the treatment of advanced GISTs, the majority of patients develop resistance to imatinib within 2 to 3 years. Even if acquired mutations in KIT or PDGFRA represent the most frequent event underlying secondary resistance, a signaling crosstalk between KIT and FGFR has been suggested as an alternative mechanism promoting resistance to imatinib [24]. Interestingly, FGFR3 silencing restored imatinib response in imatinib-resistant GIST cells, whereas the addition of FGF2 to GIST cells induced KIT signaling, increased cell proliferation and desensitized GIST cells to imatinib, suggesting the existence of a crosstalk between the two receptors. Notably, the authors also found that rescue of cell growth by FGF2 was ineffective after KIT knockdown, and FGF2-induced imatinib resistance was inhibited by FGFR3 silencing. Moreover, increased protein levels of FGF2 were found in imatinib-resistant with respect to imatinib-sensitive GIST cells, a finding that was also confirmed in GIST patient samples [24]. Similarly, Li et al. have confirmed that FGF2 rescues the effect of KIT inhibition and reduces the sensitivity of imatinib in GIST cells, and conversely, the antiproliferative activity of imatinib was enhanced by cotreatment with BGJ398, a potent FGFR1–3 inhibitor, alone or in the presence of added FGF2 [25].
The crosstalk between KIT and FGFR in playing an important role in imatinib resistance has been also extensively studied by Boichuck and co-workers [26][27][28][29]. In fact, they found that FGFR inhibition reduced cellular viability, induced apoptosis, and affected the growth kinetics of imatinib-resistant GIST cells in vitro, while inhibition of FGF-signaling restored the susceptibility to imatinib in resistant GIST cells [28]. Moreover, the authors tested the efficacy of BGJ398 in xenograft models of GISTs exhibiting secondary imatinib resistance due to loss of KIT/gain of FGFR2, showing that inhibition of FGFR signaling combined with imatinib treatment had a striking effect on tumor growth in vivo.
6. Clinical Perspectives
Given that the deregulation of FGFR signaling has been observed in a subset of many cancers, in the last decade, several multi-target compounds that inhibit vascular endothelial growth factor and PDGF receptors, in addition to FGFRs, have been developed. Many such inhibitors, both multi-target as well as FGFR-specific tyrosine kinase inhibitors (TKIs), monoclonal antibodies, and FGF ligand traps are already in clinical trials or already approved for the treatment of many cancers.
Currently regorafenib, an oral multi-kinase inhibitor that blocks the activity of multiple protein kinases, including FGFR, is the only one approved in metastatic-resistant GISTs, after failure of imatinib and sunitinib [30]. Notably, it has been shown that, together with patients carrying the KIT exon 11 altogether with exon 17 activation loop resistance mutation, SDH-deficient GISTs experienced greater clinical benefit from regorafenib, suggesting that this subtype may be more addicted to FGF/FGFR signaling [31]. According to this biological rationale, the ongoing REGISTRI trial is evaluating if KIT/PDGFRA WT may benefit from regorafenib upfront in first line.
Beside regorafenib, in the last decade other multi-TKIs active also against FGFR, such as dovitinib, masitinib, ponatinib and pazopanib, have been tested in GISTs.
The recent accumulating evidence of the specific role of the FGF/FGFR pathway in almost all molecular subgroups of GIST patients has raised much more interest in the evaluation of selective FGFR inhibitors, besides the known multi-TKIs. However, the only available results came from a single-center, phase IB study, evaluating the combination of BGJ398, a selective FGFR1–4 inhibitor, with imatinib in patients with imatinib-refractory advanced GISTs [31]. Clinical response was limited, with stable disease as the best response observed in seven patients by RECIST 1.1, and a median PFS of 12.1 weeks with the combination therapy, that is anyway superior to that achieved with imatinib re-challenge [31]. The study population included a series of heavily pre-treated KIT-mutant patients, irrespective of molecular-based selection, and the high toxicity encountered with the drug combination does not allow to draw conclusions on specific molecular subtypes’ response to FGFR inhibition.
7. Conclusions
Taken together, all these findings show that the FGF/FGFR pathway activation may play a role in the pathogenesis of specific molecular subsets of GISTs, therefore its inhibition may have some windows of opportunity also in GIST management.
References
- Miettinen, M.; Wang, Z.-F.; Sarlomo-Rikala, M.; Osuch, C.; Rutkowski, P.; Lasota, J. Succinate Dehydrogenase-Deficient GISTs. Am. J. Surg. Pathol. 2011, 35, 1712–1721.
- Boikos, S.A.; Pappo, A.S.; Killian, J.K.; LaQuaglia, M.P.; Weldon, C.B.; George, S.; Trent, J.C.; von Mehren, M.; Wright, J.A.; Schiffman, J.D.; et al. Molecular Subtypes of KIT/PDGFRA Wild-Type Gastrointestinal Stromal Tumors. Jama Oncol. 2016, 2, 922.
- Pantaleo, M.A.; Astolfi, A.; Urbini, M.; Nannini, M.; Paterini, P.; Indio, V.; Saponara, M.; Formica, S.; Ceccarelli, C.; Casadio, R.; et al. Analysis of all subunits, SDHA, SDHB, SDHC, SDHD, of the succinate dehydrogenase complex in KIT/PDGFRA wild-type GIST. Eur. J. Hum. Genet. 2014, 22, 32–39.
- Miettinen, M.; Fetsch, J.F.; Sobin, L.H.; Lasota, J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: A clinicopathologic and molecular genetic study of 45 cases. Am. J. Surg. Pathol. 2006, 30, 90–96.
- Daniels, M.; Lurkin, I.; Pauli, R.; Erbstößer, E.; Hildebrandt, U.; Hellwig, K.; Zschille, U.; Lüders, P.; Krüger, G.; Knolle, J.; et al. Spectrum of KIT/PDGFRA/BRAF mutations and Phosphatidylinositol-3-Kinase pathway gene alterations in gastrointestinal stromal tumors (GIST). Cancer Lett. 2011, 312, 43–54.
- Gasparotto, D.; Rossi, S.; Polano, M.; Tamborini, E.; Lorenzetto, E.; Sbaraglia, M.; Mondello, A.; Massani, M.; Lamon, S.; Bracci, R.; et al. Quadruple-Negative GIST Is a Sentinel for Unrecognized Neurofibromatosis Type 1 Syndrome. Clin. Cancer Res. 2017, 23, 273–282.
- Pantaleo, M.A.; Nannini, M.; Corless, C.L.; Heinrich, M.C. Quadruple wild-type (WT) GIST: Defining the subset of GIST that lacks abnormalities of KIT, PDGFRA, SDH, or RAS signaling pathways. Cancer Med. 2015, 4, 101–103.
- Nannini, M.; Astolfi, A.; Urbini, M.; Indio, V.; Santini, D.; Heinrich, M.C.; Corless, C.L.; Ceccarelli, C.; Saponara, M.; Mandrioli, A.; et al. Integrated genomic study of quadruple-WT GIST (KIT/PDGFRA/SDH/RAS pathway wild-type GIST). BMC Cancer 2014, 14, 685.
- Nannini, M.; Urbini, M.; Astolfi, A.; Biasco, G.; Pantaleo, M.A. The progressive fragmentation of the KIT/PDGFRA wild-type (WT) gastrointestinal stromal tumors (GIST). J. Transl. Med. 2017, 15, 113.
- Brenca, M.; Rossi, S.; Polano, M.; Gasparotto, D.; Zanatta, L.; Racanelli, D.; Valori, L.; Lamon, S.; Dei Tos, A.P.; Maestro, R. Transcriptome sequencing identifies ETV6-NTRK3 as a gene fusion involved in GIST. J. Pathol. 2016, 238, 543–549.
- Shi, E.; Chmielecki, J.; Tang, C.M.; Wang, K.; Heinrich, M.C.; Kang, G.; Corless, C.L.; Hong, D.; Fero, K.E.; Murphy, J.D.; et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J. Transl. Med. 2016, 14, 339.
- Pantaleo, M.A.; Urbini, M.; Indio, V.; Ravegnini, G.; Nannini, M.; de Luca, M.; Tarantino, G.; Angelini, S.; Gronchi, A.; Vincenzi, B.; et al. Genome-wide analysis identifies MEN1 and MAX mutations and a neuroendocrine-like molecular heterogeneity in Quadruple WT GIST. Mol. Cancer Res. 2017, 15, 553–562.
- Itoh, N.; Ornitz, D.M. Fibroblast growth factors: From molecular evolution to roles in development, metabolism and disease. J. Biochem. 2011, 149, 121–130.
- Itoh, N.; Ornitz, D.M. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004, 20, 563–569.
- Trueb, B. Biology of FGFRL1, the fifth fibroblast growth factor receptor. Cell. Mol. Life Sci. 2011, 68, 951–964.
- Kalinina, J.; Dutta, K.; Ilghari, D.; Beenken, A.; Goetz, R.; Eliseenkova, A.V.; Cowburn, D.; Mohammadi, M. The alternatively spliced acid box region plays a key role in FGF receptor autoinhibition. Structure 2012, 20, 77–88.
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129.
- Ornitz, D.M. FGFs, heparan sulfate and FGFRs: Complex interactions essential for development. BioEssays 2000, 22, 108–112.
- Ornitz, D.M.; Itoh, N. The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266.
- Hallinan, N.; Finn, S.; Cuffe, S.; Rafee, S.; O’Byrne, K.; Gately, K. Targeting the fibroblast growth factor receptor family in cancer. Cancer Treat. Rev. 2016, 46, 51–62.
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332.
- Urbini, M.; Indio, V.; Tarantino, G.; Ravegnini, G.; Angelini, S.; Nannini, M.; Saponara, M.; Santini, D.; Ceccarelli, C.; Fiorentino, M.; et al. Gain of FGF4 is a frequent event in KIT/PDGFRA/SDH/RAS-P WT GIST. Genes Chromosomes Cancer 2019, 58, 636–642.
- Flavahan, W.A.; Drier, Y.; Johnstone, S.E.; Hemming, M.L.; Tarjan, D.R.; Hegazi, E.; Shareef, S.J.; Javed, N.M.; Raut, C.P.; Eschle, B.K.; et al. Altered chromosomal topology drives oncogenic programs in SDH-deficient GISTs. Nature 2019, 575, 229–233.
- Javidi-Sharifi, N.; Traer, E.; Martinez, J.; Gupta, A.; Taguchi, T.; Dunlap, J.; Heinrich, M.C.; Corless, C.L.; Rubin, B.P.; Druker, B.J.; et al. Crosstalk between KIT and FGFR3 promotes gastrointestinal stromal tumor cell growth and drug resistance. Cancer Res. 2015, 75, 880–891.
- Li, F.; Hung, H.; Li, X.; Ruddy, D.A.; Wang, Y.; Ong, R.; Chow, P.; Qiu, S.; Tam, A.; Rakiec, D.P.; et al. FGFR-mediated reactivation of MAPK signaling attenuates antitumor effects of imatinib in gastrointestinal stromal tumors. Cancer Discov. 2015, 5, 438–451.
- Boichuk, S.; Dunaev, P.; Galembikova, A.; Mustafin, I.; Valeeva, E. Inhibition of fibroblast growth factor receptor-signaling sensitizes imatinib-resistant gastrointestinal stromal tumors to low doses of topoisomerase II inhibitors. Anti-Cancer Drugs 2018, 29, 549–559.
- Boichuk, S.; Dunaev, P.; Galembikova, A.; Bikinieva, F.; Nurgatina, I.; Mustafin, I.; Aukhadieva, A.; Kurtasanov, R.; Andriutsa, N.; Shagimardanova, E.; et al. Inhibition of FGFR2-signaling attenuates a homology-mediated dna repair in gist and sensitizes them to DNA-topoisomerase II inhibitors. Int. J. Mol. Sci. 2020, 21, 352.
- Boichuk, S.; Galembikova, A.; Dunaev, P.; Valeeva, E.; Shagimardanova, E.; Gusev, O.; Khaiboullina, S. A novel receptor tyrosine kinase switch promotes gastrointestinal stromal tumor drug resistance. Molecules 2017, 22, 2152.
- Boichuk, S.; Galembikova, A.; Dunaev, P.; Micheeva, E.; Valeeva, E.; Novikova, M.; Khromova, N.; Kopnin, P. Targeting of FGF-Signaling Re-Sensitizes Gastrointestinal Stromal Tumors (GIST) to Imatinib in Vitro and in Vivo. Molecules 2018, 23, 2643.
- Demetri, G.D.; Reichardt, P.; Kang, Y.K.; Blay, J.Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302.
- Ben-Ami, E.; Barysauskas, C.M.; von Mehren, M.; Heinrich, M.C.; Corless, C.L.; Butrynski, J.E.; Morgan, J.A.; Wagner, A.J.; Choy, E.; Yap, J.T.; et al. Long-term follow-up results of the multicenter phase II trial of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of standard tyrosine kinase inhibitor therapy. Ann. Oncol. 2016, 27, 1794–1799.


