Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nina Kaminen-Ahola | + 4130 word(s) | 4130 | 2021-08-03 05:27:52 | | | |
| 2 | Camila Xu | Meta information modification | 4130 | 2021-08-03 09:57:39 | | | | |
| 3 | Camila Xu | Meta information modification | 4130 | 2021-08-03 10:01:41 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kaminen-Ahola, N. Early Prenatal Alcohol Exposure. Encyclopedia. Available online: https://encyclopedia.pub/entry/12688 (accessed on 07 February 2026).
Kaminen-Ahola N. Early Prenatal Alcohol Exposure. Encyclopedia. Available at: https://encyclopedia.pub/entry/12688. Accessed February 07, 2026.
Kaminen-Ahola, Nina. "Early Prenatal Alcohol Exposure" Encyclopedia, https://encyclopedia.pub/entry/12688 (accessed February 07, 2026).
Kaminen-Ahola, N. (2021, August 03). Early Prenatal Alcohol Exposure. In Encyclopedia. https://encyclopedia.pub/entry/12688
Kaminen-Ahola, Nina. "Early Prenatal Alcohol Exposure." Encyclopedia. Web. 03 August, 2021.
Copy Citation
Prenatal alcohol exposure (PAE) is the underlying cause for a variety of birth defects referred to as Fetal Alcohol Spectrum Disorders (FASD).
embryonic stem cells
environmental epigenetics
epigenetic reprogramming
DNA methylation
1. Introduction
1.1. Consequences of PAE
Prenatal alcohol exposure (PAE) is the underlying cause for a variety of birth defects referred to as Fetal Alcohol Spectrum Disorders (FASD). It is a non-diagnostic umbrella term for all the alcohol-related developmental disorders and birth defects, including four diagnostics categories [1]. The most severe form is known as fetal alcohol syndrome (FAS) with diagnosed growth retardation, central nervous system neurodevelopmental deficits, and craniofacial dysmorphology [2][3][4]. After the identification of FAS, it became clear that not all prenatally alcohol exposed individuals expressed all the features of FAS. Alcohol-associated disabilities represent a spectrum, consisting of growth deficits from mild to severe, physical abnormalities, neurocognitive, and behavioral deficits, as well as an increased vulnerability to mental health problems and other comorbidities [5][6][7]. In addition to FAS, there are the following three other diagnostic categories: partial fetal alcohol syndrome (PFAS), alcohol-related neurodevelopmental disorders (ARND), and alcohol-related birth defects (ARBD) [1][2]. PAE is a substantial cause of mental disability and birth defects in the Western world with an estimated 3–5% prevalence in Europe and North America and over 10% prevalence in South Africa [8]. Several biological and environmental factors are known to influence the effects of alcohol exposure, including the dose of alcohol, the exposure pattern, the duration of exposure, the developmental timing of exposure, the genetic background of the mother and fetus, maternal age, socioeconomic status, nutrition, and interactions with other drugs. Owing to these factors, the phenotype of FASD is complex and the molecular mechanisms of PAE are challenging to study.
The growing amount of evidence suggests that some developmental periods are particularly sensitive to alcohol teratogenesis [9][10]. The preimplantation period lasts for the first two weeks of gestation in humans and 4–6 gestational days (GD) in mice. This period is vulnerable to alcohol exposure [11][12] since the embryo is undergoing rapid developmental changes as the zygote develops into a morula and further into a blastocyst. After the preimplantation period, the blastocyst attaches to the uterine wall and gastrulation begins, which is a time of intense cell differentiation into three germ layers—endoderm, mesoderm, and ectoderm. This period occurs during week three in humans and GD6.5–8.5 in mice. The differentiating cells are particularly sensitive to alcohol exposure, which makes gastrulation one of the most vulnerable developmental periods [13][14][15]. Studies have shown that a single dose of alcohol on any of GD7–9 can produce different craniofacial malformations resembling the features of FASD in mice [16][17][18]. In addition, differences as small as 4 h in the timing of PAE on GD7 resulted in different craniofacial phenotypes [19][20]. Gastrulation is a part of organogenesis, which corresponds to 3–8 weeks of gestation in humans and GD7‒14 in mice. It is a period of continuing cell differentiation and the beginning of rudimentary organ formation, and, therefore, alcohol exposure during organogenesis may produce many features of FASD [21]. These studies have shown that early pregnancy is a sensitive time for alcohol-induced developmental disruptions. Here, we define early pregnancy as the period from fertilization to organogenesis, i.e., weeks 0–8 of gestation in humans and GD0–14 in mice.
1.2. Effects of Early Environmental Exposures
Numerous studies have shown that environmental exposures in early pregnancy, and even before pregnancy, can have an impact not only on development but also permanently on a person’s overall health and risk for adult diseases such as cancer, heart disease, obesity, or diabetes [22][23][24][25][26]. David Barker proposed a hypothesis of “Developmental Origins of Adult Health and Disease” (DOHaD), suggesting that the early environment programs fetal development and permanently affects the physiology of the fetus [27]. Although the significant role of the prenatal environment in an adult phenotype seems to be indisputable, the molecular mechanisms underlying these associations or causality of the observed alterations are poorly understood.
The epigenetic variation induced during in utero development has become a strong candidate mediator of the environmental effects. Prenatal exposure to various environmental factors, such as chemicals [28], diet [29], stress [30], obesity [31], tobacco smoking [32], and alcohol consumption [33] has been shown to alter epigenetic modifications in the human and mouse offspring, leading to potential consequences in the phenotype.
The vulnerability of the epigenome in early pregnancy is well demonstrated in studies of prenatal famine and bisphenol A (BPA) exposures. After maternal exposure to famine during the “Hunger winter” in 1944–1945 in the Netherlands, children had a higher risk of having obesity, heart diseases, and diabetes in adulthood [24]. The famine exposure was associated with a decreased DNA methylation of growth promoting the insulin-like growth factor 2 (IGF2) imprinted gene in the whole blood of individuals exposed particularly at early pregnancy [34]. In further studies, DNA methylation changes specific to early pregnancy were also detected in other imprinted genes that are involved in growth as well as in metabolic and cardiovascular disease [35]. The other example, BPA exposure, has been linked to DNA methylation changes and genomic imprinting disruption in prenatally exposed mice [28][36]. Maternal prenatal BPA exposure during the late stages of mouse oocyte development and the early stages of embryonic development (maternal exposure 2 weeks prior to mating until GD9.5 and GD12.5) significantly altered embryonic and placental DNA methylation and the expression of imprinted genes associated with imprinting disorders in humans [37]. In contrast, exposure in later stages of development had no significant effects on the imprinted genes (maternal exposure GD5.5–GD12.5).
The earliest stages of pregnancy and embryonic development comprise a sensitive period of epigenetic reprogramming (Figure 1). During reprogramming, epigenetic marks, especially DNA methylation, are erased and re-established in the early embryo. It provides a regulatory mechanism for the differentiation of cells toward different cell types—reprogramming restores the ability of the zygote to develop into all the different cell types and tissues [38][39]. Environmental-induced disruptions during this period may influence the regulation of developmental genes, resulting in alterations of embryonic development and adverse health outcomes.
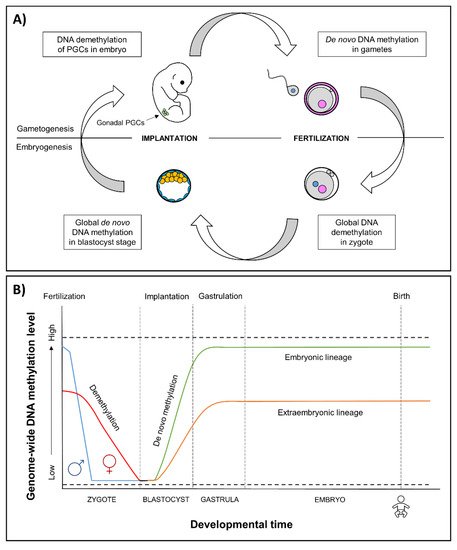
Figure 1. (A) Epigenetic reprogramming in the germ line and in somatic cells. Reprogramming occurs in two phases of in utero development, one after fertilization in the preimplantation embryo and the other in the developing gametes of the fetus. In gametogenesis, primordial germ cells (PGCs) become globally demethylated early in development [40]. De novo methylation begins in prospermatogonia in male germ cells during spermatogenesis and after birth in maturing oocytes. In embryogenesis, the epigenetic reprogramming begins after fertilization and continues until the blastocyst stage. Genome-wide de novo methylation occurs during implantation and gastrulation, leading to the formation of different cell types. (B) Epigenetic reprogramming in embryos. After fertilization, in the beginning of reprogramming, methyl marks are actively erased in paternal genome (blue line), and passively erased in maternal genome (red line) [41]. By the blastocyst stage, the genome is almost completely hypomethylated [42]. Blastocyst cells divide and differentiate into the following two distinct lineages: the pluripotent inner cell mass, which will become the developing fetus, and trophoectoderm, which forms the extraembryonic tissues. Demethylation is followed by genome-wide de novo methylation during implantation and gastrulation [39], in which the reestablishment of epigenetic marks is more limited in the extraembryonic lineage (orange line) compared to embryonic lineage (green line) [43]. An exception to methylation is made by imprinted genes that remain either methylated or unmethylated throughout epigenetic reprogramming (dashed lines) [39].
2. Epigenetic Effects of Early PAE
Several human and animal studies have identified associations between PAE and alterations in epigenetic modifications such as DNA methylation, histone modifications, and non-coding RNAs, including microRNAs (miRNAs) [44][45][46]. However, the molecular mechanisms underlying these alcohol-induced epigenetic effects are still poorly understood. It has been suggested that early PAE may interfere with epigenetic reprogramming and could change the establishment of epigenetic marks. One suggested mechanism is the alcohol-induced reduction in folate, B6, or B12 vitamins in the methionine cycle, which decreases the amount of S-adenosylmethionine (SAM) in the cells [47][48][49]. As SAM is a donor of methyl groups for both DNA and histone methylation, alcohol could change the establishment of epigenetic marks in a developing embryo. Furthermore, acetyl groups from alcohol were rapidly incorporated in histone acetylation in the brain in a mouse model and changes have been observed also in a prenatally alcohol exposed fetal brain [46]. Epigenetic changes in the first embryonic cells could be fixed in persistent cellular memory and mitotically transmitted to different cell and tissue types. Consequently, these alterations may affect gene regulation and depending on the function of the cell types or tissues, they will contribute to the complex phenotype of FASD.
2.1. Effects of Early PAE on Epigenome in Mouse Models
Only a few studies have examined the epigenetic effects of in vivo alcohol exposure during the first trimester thus far. The first global methylation study was performed by Garro et al. [50]. Acute alcohol administration twice a day during GD9–11 (in total of five 3 g/kg doses by gavage) resulted in global hypomethylation in DNA methylation profiles in mouse fetuses, potentially by inhibiting DNA methyltransferase 1 (Dnmt1) activity. In later studies, early PAE-induced epigenetic changes were detected in the placenta, but the embryos remained unaffected. Haycock and Ramsay [51] studied the effects of alcohol in GD10.5 mouse embryos and placentas exposed during preimplantation development (2.9 g/kg ethanol injections in females on GD1.5 and GD2.5) and found decreased DNA methylation at the imprinting control region of the Igf2/H19-imprinted locus in the placentas.
By using a mouse whole-embryo culture, Liu et al. [52] investigated the effects of PAE (88 mM ethanol exposure on GD8.5 throughout 44 h) at early embryonic neurulation. They showed that early PAE causes changes in DNA methylation with associated changes in gene expression and found significant methylation changes in imprinted genes and genes known to have roles in growth, cell cycle, apoptosis, cancer, and olfaction. In this study, specific regions became less methylated and others more methylated in response to alcohol exposure, suggesting that some regions may be more sensitive to the effects of alcohol-induced alterations. In addition, they observed delayed growth and reduced overall growth with significant alteration in the development of the heart, caudal neural tube, brain vesicles, optic system, and limb buds of the embryos treated with alcohol.
By using our mouse model of early PAE (maternal ad libitum ingestion of 10% (v/v) ethanol during GD0.5–8.5, approximately 12 g/kg/day), we have shown for the first time that alcohol could affect the adult phenotype by altering the epigenotype of the early mouse embryo [53]. We studied the effects of early PAE by using a metastable epiallele Agouti viable yellow (Avy), which is a dominant mutation of the murine Agouti (A) locus, caused by the insertion of an intracisternal A-particle (IAP) retrotransposon upstream of the Agouti coding exons. The activity of Avy is variable among genetically identical mice, resulting in mice with a range of coat colors; from yellow to mottled to agouti (termed pseudoagouti) [54]. Avy is a widely used “biosensor” to study the effects of environmental exposures on the epigenome [55][56]. Our results demonstrated that early PAE increases the DNA methylation level at the Avy allele and, consequently, alters the coat color of offspring [53]. Furthermore, we found similar changes in gene expression in the hippocampus, olfactory epithelium, and mesodermal bone marrow of adolescent mice, suggesting that changes in gene regulation may have already occurred in the first cells of the embryo [57].
2.2. Embryonic Stem Cells as a Model for Early Alcohol-Induced Effects
Most of the early alcohol exposure studies have focused on exposure in in vitro models using undifferentiated or differentiating stem cells. Embryonic stem cells (ESCs) derived from the inner cell mass of a developing blastocyst are pluripotent, having the developmental potential to give rise to all three embryonic germ layers and eventually differentiate into all cells and tissues of an adult organism. The pluripotent identity of ESCs is governed by a network of transcriptional factors, signaling pathways, epigenetic regulators, and structurally open chromatin that holds ESCs in an undifferentiated state. In response to differentiation signals, the transcriptome, epigenome, and chromatin structure in the differentiating cells undergo rapid global changes that silence the pluripotency genes and activate selected lineage-specific genes [58][59].
Since differentiation can be experimentally induced in vitro, and given their indefinite self-renewing capacity, ESCs are a useful model to study cellular identity and early developmental events during embryogenesis. Their environment can be easily manipulated, which makes them a valuable tool to clarify the interaction between genes and environment in the beginning of embryonic development. ESCs have been used in toxicological research and several human and mouse stem cell models have been established to study the early effects of alcohol. As alcohol strongly disrupts the development of the nervous system [60], several models of neural differentiation have also been developed to study the effects of alcohol on early human neural and brain development. To date, studies on ESCs have observed alterations in the proliferation and differentiation properties as well as changes in molecular mechanisms such as in the levels of core pluripotency factors and epigenetic marks [61][62].
2.3. Effects of Alcohol on Embryonic Stem Cells and Differentiation
Studies on mouse (mESC) and human (hESC) embryonic stem cells have shown that differentiating cells and developing tissues are more vulnerable to alcohol than already differentiated cells. Arzumnayan et al. [13] showed that 80–84 mM ethanol exposure for 1–6 days affected neither the proliferation nor the expression of pluripotency markers of undifferentiated mESCs, but triggered apoptosis during embryonic body (EB) differentiation. Nash et al. [63], in turn, showed that a low dose of ethanol (20 mM for one week) increases cell proliferation and induces larger colonies, and simultaneously increases cell apoptosis in undifferentiated cells and ethanol-exposed hESC-derived neural progenitor cells. Moreover, Taléns-Visconti et al. [64] showed that ethanol exposure (25 and 50 mM in proliferating or differentiating media) not only impairs neural progenitor cell survival, but also the differentiation of hESCs into neural progenitors and further into mature neurons and astrocytes. Ethanol exposure also induced expression changes of neural differentiation-associated genes and disrupted the actin cytoskeleton of neural progenitors [64].
There is only scarce evidence of the molecular mechanisms behind the alcohol-induced effects on cell differentiation. One potential mechanism is the effects on the signaling pathways that have important roles in the regulation of stem cell differentiation and the control of embryonic development, such as the WNT pathway [65]. Ethanol exposure (25, 50, and 100 mM after definitive endoderm stage until harvesting) has been shown to suppress the early hepatic differentiation of hESC-derived hepatic progenitor cells in a dose-dependent manner by inhibiting WNT as well as the MAPK/ERK pathway [66]. Furthermore, alcohol-induced inhibition in WNT signaling was also observed during human neural stem cell (NSC) differentiation [67] and the cardiac differentiation of mESCs [68][69].
Another possible underlying mechanism of the alcohol-induced effects on differentiation is alterations in the levels of core pluripotency factors POU class 5 homeobox 1 (Oct4), SRY-box transcription factor 2 (Sox2), and Nanog Homeobox (Nanog) that control the pluripotent state of ESCs [70]. In mESCs, ethanol exposure has been seen to inhibit the loss of core pluripotency markers during both EB (81–84 mM ethanol for 1–6 days) [13] and cardiac differentiation (17.1–51.4 mM for 14 days) [68], suggesting that alcohol may delay the ESC differentiation.
In addition to maintaining pluripotency, the core pluripotency factors are involved in lineage selection during early differentiation events. The dosage of Oct4 and Sox2 in differentiating cells determines the lineage commitment—an increased Oct4/Sox2 ratio induces ESCs to differentiate into mesoendoderm (ME) and, by contrast, a decreased Oct4/Sox2 ratio into neuroectoderm (NE) [71][72][73]. Interestingly, alcohol has been shown to affect the balance of these lineage specifiers and alter the differentiation trajectory into specific lineages, especially when differentiating into NE. Ogony et al. [74] found that ethanol exposure (25, 50, and 100 mM for 0–6 days) increases the expression of Oct4 in a dose- and time-dependent manner, elevates the overall Oct4/Sox2 ratio, and misleads the cells into an ME cell fate during ESC differentiation into NE. Furthermore, they investigated the effects of ethanol (100 mM for 0–6 days) on the Oct4/Sox2 ratio as well as on the expression of other downstream genes involved in pluripotency, differentiation, and signaling during the mESC differentiation into NE [62]. Alcohol exposure was shown to downregulate 19 pluripotency genes and upregulate 14 differentiation-associated genes. The changes in the NE differentiation-associated genes altered the overall gene expression dynamics, which could explain the different trajectory observed during the differentiation of alcohol-exposed cells [62].
These results collectively suggest that alcohol reprograms the lineage specification by changing the balance of core pluripotency factors Oct4 and Sox2, thereby forcing ESCs away from neuroectodermal cell fate. This is in line with previous animal studies in which alcohol exposure during gastrulation caused neural progenitor pool reduction [15] as well as long-term effects on the forebrain and mature brain stem nuclei structures [75]. The effect of alcohol on lineage specification is well illustrated by Waddington’s epigenetic landscape model, which describes how development is unidirectional, meaning that embryonic stem cells develop into a mature differentiated state [76]. If alcohol disrupts the balance of the core pluripotency factors, it could force stem cells down the differentiation trajectory away from neuroectoderm (Figure 2). This misguidance could result in developmental delay and defects of the nervous system underlying FASD phenotypes.
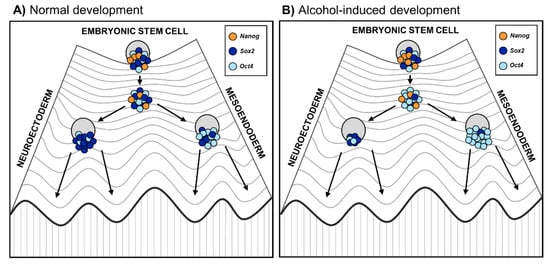
Figure 2. Embryonic stem cell differentiation into germ layers during (A) normal embryonic development and (B) altered development due to prenatal alcohol exposure. The Waddington’s epigenetic landscape reflects the differentiation of cells, where the fate of stem cells depends on their history as well as developmental and environmental inputs [76]. On the highest hill are ESC progenies that progress toward differentiation. Changes in epigenetic modifications in ESC progenies lead to a reconfiguration of the core transcription factors Oct4, Sox2, and Nanog, represented as a Waddington’s landscape [77]. Coming down to the hill, stem cells first reach a plateau with low Nanog expression that enables them to respond to differentiation signals. (A) During normal stem cell differentiation, higher Oct4 expression induces stem cells toward the mesoendoderm progenitor cell fate, while higher Sox2 drives stem cells toward the neuroectoderm progenitor cell fate; (B) During alcohol-altered stem cell differentiation, alcohol affects the balance of the core pluripotency factors, especially Oct4/Sox2 ratio [74]. Therefore, alcohol may reprogram lineage specification favoring excess of Oct4 relative to Sox2 and forcing stem cells down the differentiation trajectory away from neuroectoderm.
2.4. Epigenetic Effects of Alcohol on Embryonic Stem Cells and Differentiation
The effects of alcohol on the epigenome have been mainly studied in differentiated cells such as NSCs and heart progenitors rather than in undifferentiated ESCs or specifically in gastrulation. However, the results have shown associations between ethanol exposure and alterations in epigenetic mechanisms.
Alcohol-induced changes in the DNA methylation of ESCs and differentiating cells were studied in a genome-wide DNA methylation sequencing analysis, which revealed significant alterations in the methylation and transcriptomic profiles of ethanol-treated (20 mM for 24 h) undifferentiated hESCs, leading to reduced pluripotency, and also ethanol-treated differentiated EBs [61]. A higher global hypermethylation in undifferentiated ESCs than in EBs was observed at the promoter regions, suggesting that the methylomes of undifferentiated ESCs are more prone to alcohol-induced effects than the methylomes of already differentiated cells. The changes in transcriptomic of undifferentiated hESCs and EBs were associated with oxidative stress, metabolic processes, and neuronal properties [61].
DNA methylation changes have also been detected in ethanol-exposed mouse NSCs. Hicks et al. [78] focused on promoter regions and found that ethanol exposure (86.8 mM for 48 h) prolonged the total length of the cell cycle, increased the activity of Dnmt1 and induced hypermethylation of several cell cycle genes. Considerably, the results of the increased activity of Dnmt1 in NSCs contradict the findings of Garro et al. [50], who showed that PAE decreases Dnmt1 activity in fetal mice. Moreover, Zhou et al. [79] found that binge-like ethanol exposure (88 mM for 6 h) delayed the migration, neuronal formation, and growth of rat NSCs and prevented the methylation of genes associated with neural development, eye development, and developmental disorders during the reprogramming of quiescent NSCs into differentiation. Ethanol exposure (70 mM for 48 h or 8 days) during NCS differentiation has also been shown to increase the expression of methyl CpG binding protein 2 (Mecp2), an important epigenetic factor in the brain, in association with decreased DNA methylation and increased hydroxymethylation at its regulatory elements [80].
The knowledge of alcohol´s effects on the chromatin structure in ESCs is scant, but a few studies show alterations of histone modifications in NSCs [81][82] and heart progenitor cells [83][84]. Veazey et al. [81][82] studied the effects of alcohol on histone modifications (H3K4me3, H3K9me2, H3K9ac, H3K27me3) at specific genes associated with development in fetal mouse NSCs and showed that ethanol exposure (35 and 52 mM for 3 days and 70 mM for 5 days) exhibits significant alterations in the chromatin structure. The effects were dependent on the gene of interest and the dose of ethanol as well as whether the cells had recovered from the treatment before harvesting [82]. Interestingly, after the recovery, most of the changes were observed in histone marks associated with a repressive chromatin structure (H3K9me2 and H3K27me3). These changes were also consistent with the repressed chromatin structure observed in the in vivo mouse model (ethanol exposure on GD7), correlating with the development of craniofacial and central nervous system defects as well as a decreased expression of genes that are associated with development and epigenetic reprogramming [82]. The ethanol exposure (50 and 200 mM for 24 h) of mouse heart progenitor cells has been shown to increase H3K9 acetylation and change the expression of the genes involved in heart development [83][84].
Extensive alcohol-mediated changes in miRNA and miRNA-target gene expression have been observed in utero that can be detrimental to the developing embryo [85]. These alterations are mainly studied in mouse neural progenitor cells in the early embryonic stage. For instance, Sathyan et al. [86] found that ethanol (70 mM for 5 days) suppresses the expression of miRNAs (miR-21, miR-335, miR-9, and miR153) in cerebral cortical neuroepithelial precursors. Later, they reported that exposure also suppressed miR-140-3p in cultured neurospheres, suggesting that relatively small teratogen-induced changes in miRNAs during stem cell differentiation can result in long-lasting deficits in brain function [87].
3. Effects of Early PAE-Induced Epigenetic Alterations on Phenotype
Although PAE has been associated with alterations in the epigenome, the causal molecular mechanisms between the alcohol-induced epigenetic alterations and the adult phenotype are still unknown. Any epigenetic changes during early or even whole pregnancy that alter gene regulation and result in a human FASD phenotype are not yet revealed. Due to the limited number of human studies, the only evidence of a connection between early alcohol-induced epigenetic alterations and the phenotype is thus far based on studies using animal models.
The hypothesis of the early epigenetic origin of alcohol-induced disorders is supported by studies that have found epigenetic changes associated with the phenotype or comorbidities of FASD in various tissues. In GD9 mice embryos, acute PAE (5.8 g/kg, intragastric intubation) caused a subtle decrease in the DNA methylation of Igf2’s differentially methylated region in embryonic tissue, which led to a decrease in Igf2 gene expression [88]. These gene expression changes were associated with skeletal malformations that resemble those found in individuals with FAS. Moreover, studies on GD7.5–16.5 mouse embryos found that PAE (56% (v/v) ethanol by gavage) is linked, in addition, to global H3K9 hyperacetylation, also GATA binding protein 4 (Gata4) promoter histone H3K9 hyperacetylation, which leads to Gata4 overexpression in cardiac tissue [89][90]. Gata4 is an essential transcription factor during heart development, and it is presumed that alterations of Gata4 expression can affect the epigenetic regulation of embryonic development and contribute to congenital heart disease in children [90]. Subsequently, studies have shown that PAE (56% (v/v) ethanol by gavage or intragastric administration) increases the mRNA expression of developmental genes and also causes the hyperacetylation of H3K14 in the fetal hearts of mice [91][92].
Although we have shown for the first time that early PAE could affect the epigenome of an early embryo and, consequently, an adult phenotype by using the metastable epiallele Avy [53], it is not a normal allele and, as such, cannot prove a link between early epigenetic alterations, gene expression, and the phenotype. However, adult PAE offspring without Avy allele were examined and the phenotype was reminiscent of human FASD with craniofacial dysmorphology, postnatal growth restriction [93], and structural changes in the central nervous system [57][94]. Interestingly, this same mouse model, with relatively moderate PAE, is associated with decreased DNA methylation in the solute carrier family 17 (Slc17a6) promoter region, which plays a role in neurotransmission, synaptic plasticity, and cognition and encodes vesicular glutamate transporter (VGLUT2) in mice [95]. Slc17a6 showed increased mRNA levels together with decreased promoter DNA methylation, decreased VGLUT2 protein levels, and increased H3K4me3 in the alcohol-exposed (GD0.5–8.5) adult male hippocampus. Furthermore, 15 ethanol-sensitive miRNAs were found in the hippocampus, three of which (miR-135a, miR-135b, and miR-467b-5p) were also differentially expressed in serum, suggesting that serum expression could be used as a biomarker for expression levels in the hippocampus. The study indicates that PAE can cause long-term deregulation of brain DNA methylation, histone modification, and gene expression. A comprehensive battery of behavioral tests was conducted for the adult mice, and persistent and long-lasting alterations in behavior, including human FASD-phenotype-associated hyperactivity, were observed [96].
References
- Stratton, K.R.; Howe, C.J.; Battaglia, F.C. Fetal Alcohol Syndrome: Diagnosis, Epidemiology, Prevention, and Treatment; Diagnosis and Clinical Evaluation of Fetal Alcohol Syndrome; National Academy Press: Washington, DC, USA, 1996.
- Hoyme, H.E.; May, P.A.; Kalberg, W.O.; Kodituwakku, P.; Gossage, J.P.; Trujillo, P.M.; Buckley, D.G.; Miller, J.H.; Aragon, A.S.; Khaole, N.; et al. A practical clinical approach to diagnosis of fetal alcohol spectrum disorders: Clarification of the 1996 institute of medicine criteria. Pediatrics 2005, 115, 39–47.
- Jones, K.L.; Smith, D.W. Recognition of the fetal alcohol syndrome in early infancy. Lancet 1973, 302, 999–1001.
- Jones, K.L.; Smith, D.W.; Ulleland, C.N.; Streissguth, P. Pattern of malformation in offspring of chronic alcoholic mothers. Lancet 1973, 1, 1267–1271.
- O’Leary, C.M.; Nassarm, N.; Kurinczuk, J.J.; de Klerk, N.; Geelhoed, E.; Elliott, E.J.; Bower, C. Prenatal alcohol exposure and risk of birth defects. Pediatrics 2010, 126, 843–850.
- Popova, S.; Lange, S.; Shield, K.; Mihic, A.; Chudley, A.E.; Mukherjee, R.A.S.; Bekmuradov, D.; Rehm, J. Comorbidity of fetal alcohol spectrum disorder: A systematic review and meta-analysis. Lancet 2016, 387, 978–987.
- Riley, E.P.; Infante, M.A.; Warren, K.R. Fetal alcohol spectrum disorders: An overview. Neuropsychol. Rev. 2011, 21, 73–80.
- Roozen, S.; Peters, G.J.; Kok, G.; Townend, D.; Nijhuis, J.; Curfs, L. Worldwide Prevalence of Fetal Alcohol Spectrum Disorders: A Systematic Literature Review Including Meta-Analysis. Alcohol. Clin. Exp. Res. 2016, 40, 18–32.
- Armant, D.R.; Saunders, D.E. Exposure of embryonic cells to alcohol: Contrasting effects during preimplantation and postimplantation development. Semin. Perinatol. 1996, 20, 127–139.
- Parnell, S.E.; Holloway, H.T.; O’Leary-Moore, S.K.; Dehart, D.B.; Paniaqua, B.; Oguz, I.; Budin, F.; Styner, M.A.; Johnson, G.A.; Sulik, K.K. Magnetic resonance microscopy-based analyses of the neuroanatomical effects of gestational day 9 ethanol exposure in mice. Neurotoxicol. Teratol. 2013, 39, 77–83.
- Padmanabhan, R.; Hameed, M.S. Effects of acute doses of ethanol administered at pre-implantation stages on fetal development in the mouse. Drug Alcohol Depend. 1988, 22, 91–100.
- Pérez-Tito, L.; Bevilacqua, E.; Cebral, E. Peri-implantational in vivo and in vitro embryo-trophoblast development after perigestational alcohol exposure in the CD-1 mouse. Drug Chem. Toxicol. 2014, 37, 184–197.
- Arzumnayan, A.; Anni, H.; Rubin, R.; Rubin, E. Effects of ethanol on mouse embryonic stem cells. Alcohol. Clin. Exp. Res. 2009, 33, 2172–2179.
- Leach, R.E.; Stachecki, J.J.; Armant, D.R. Development of in vitro fertilized mouse embryos exposed to ethanol during the preimplantation period: Accelerated embryogenesis at subtoxic levels. Teratology 1993, 47, 57–64.
- Rubert, G.; Miñana, R.; Pascual, M.; Guerri, C. Ethanol exposure during embryogenesis decreases the radial glial progenitorpool and affects the generation of neurons and astrocytes. J. Neurosci. Res. 2006, 15, 483–496.
- Lipinski, R.J.; Hammond, P.; O’Leary-Moore, S.K.; Ament, J.J.; Pecevich, S.J.; Jiang, Y.; Budin, F.; Parnell, S.E.; Suttie, M.; Godin, E.A.; et al. Ethanol-induced face-brain dysmorphology patterns are correlative and exposure-stage dependent. PLoS ONE 2012, 7, e43067.
- Parnell, S.E.; O’Leary-Moore, S.K.; Godin, E.A.; Dehart, D.B.; Johnson, B.W.; Allan Johnson, G.; Styner, M.A.; Sulik, K.K. Magnetic resonance microscopy defines ethanol-induced brain abnormalities in prenatal mice: Effects of acute insult on gestational day 8. Alcohol. Clin. Exp. Res. 2009, 33, 1001–1011.
- Sulik, K.K. Genesis of alcohol-induced craniofacial dysmorphism. Exp. Biol. Med. 2005, 230, 366–375.
- Godin, E.A.; O’Leary-Moore, S.K.; Khan, A.A.; Parnell, S.E.; Ament, J.J.; Dehart, D.B.; Johnson, B.W.; Allan Johnson, G.; Styner, M.A.; Sulik, K.K. Magnetic resonance microscopy defines ethanol-induced brain abnormalities in prenatal mice: Effects of acute insult on gestational day 7. Alcohol. Clin. Exp. Res. 2010, 34, 98–111.
- Sulik, K.K.; Johnston, M.C. Sequence of developmental alterations following acute ethanol exposure in mice: Craniofacial features of the fetal alcohol syndrome. Am. J. Anat. 1983, 166, 257–269.
- Webster, W.S.; Walsh, D.A.; McEwen, S.E.; Lipson, A.H. Some teratogenic properties of ethanol and acetaldehyde in C57BL/6J mice: Implications for the study of the fetal alcohol syndrome. Teratology 1983, 27, 231–243.
- Barker, D.J. The developmental origins of chronic adult disease. Acta Paediatr. Suppl. 2004, 93, 26–33.
- Gluckman, P.D.; Hanson, M.A.; Beedle, A.S. Early life events and their consequences for later disease: A life history and evolutionary perspective. Am. J. Hum. Biol. 2007, 19, 1–19.
- Painter, R.C.; Roseboom, T.J.; Bleker, O.P. Prenatal exposure to the Dutch famine and disease in later life: An overview. Reprod. Toxicol. 2005, 20, 345–352.
- Sales, V.M.; Ferguson-Smith, A.C.; Patti, M.E. Epigenetic Mechanisms of Transmission of Metabolic Disease across Generations. Cell Metab. 2017, 25, 559–571.
- Yajnik, C.S. Early life origins of insulin resistance and type 2 diabetes in India and other Asian countries. J. Nutr. 2004, 134, 205–210.
- Hales, C.N.; Barker, D.J. Type 2 (non-insulin-dependent) diabetes mellitus: The thrifty phenotype hypothesis. Diabetologia 1992, 35, 595–601.
- Dolinoy, D.C.; Huang, D.; Jirtle, R.L. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc. Natl. Acad. Sci. USA 2007, 104, 13056–13061.
- Anderson, O.S.; Sant, K.E.; Dolinoy, D.C. Nutrition and epigenetics: An interplay of dietary methyl donors, one-carbon metabolism and DNA methylation. J. Nutr. Biochem. 2012, 23, 853–859.
- Cao-Lei, L.; de Rooij, S.R.; King, S.; Matthews, S.G.; Metz, G.A.S.; Roseboom, T.J.; Szyf, M. Prenatal stress and epigenetics. Neurosci. Biobehav. Rev. 2020, 117, 198–210.
- van Dijk, S.J.; Molloy, P.L.; Varinli, H.; Morrison, J.L.; Muhlhausler, B.S.; Members of EpiSCOPE. Epigenetics and human obesity. Int. J. Obes. 2015, 39, 85–97.
- Breton, C.V.; Byun, H.M.; Wenten, M.; Pan, F.; Yang, A.; Gilliland, F.D. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am. J. Respir. Crit. Care Med. 2009, 180, 462–467.
- Kaminen-Ahola, N. Fetal alcohol spectrum disorders: Genetic and epigenetic mechanisms. Prenat. Diagn. 2020, 40, 1185–1192.
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049.
- Tobi, E.W.; Lumey, L.H.; Talens, R.P.; Kremer, D.; Putter, H.; Stein, A.D.; Slagboom, P.E.; Heijmans, B.T. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum. Mol. Genet. 2009, 18, 4046–4053.
- Bromer, J.G.; Zhou, Y.; Taylor, M.B.; Doherty, L.; Taylor, H.S. Bisphenol-A exposure in utero leads to epigenetic alterations in the developmental programming of uterine estrogen response. FASEB J. 2010, 24, 2273–2280.
- Susiarjo, M.; Sasson, I.; Mesaros, C.; Bartolomei, M.S. Bisphenol a exposure disrupts genomic imprinting in the mouse. PLoS Genet. 2013, 9, e1003401.
- Messerschmidt, D.M.; Knowles, B.B.; Solter, D. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 2014, 28, 812–828.
- Reik, W.; Dean, W.; Walter, J. Epigenetic reprogramming in mammalian development. Science 2001, 293, 1089–1093.
- Hajkova, P.; Erhardt, S.; Lane, N.; Haaf, T.; El-Maarri, O.; Reik, W.; Walter, J.; Surani, M.A. Epigenetic reprogramming in mouse primordial germ cells. Mech. Dev. 2002, 117, 15–23.
- Mayer, W.; Niveleau, A.; Walter, J.; Fundele, R.; Haaf, T. Demethylation of the zygotic paternal genome. Nature 2000, 403, 501–502.
- Guo, H.; Zhu, P.; Yan, L.; Li, R.; Hu, B.; Lian, Y.; Yan, J.; Ren, X.; Lin, S.; Li, J.; et al. The DNA methylation landscape of human early embryos. Nature 2014, 511, 606–610.
- Chapman, V.; Forrester, L.; Sanford, J.; Hastie, N.; Rossant, J. Cell lineage-specific undermethylation of mouse repetitive DNA. Nature 1984, 307, 284–286.
- Cobben, J.M.; Krzyzewska, I.M.; Venema, A.; Mul, A.N.; Polstra, A.; Postma, A.V.; Smigiel, R.; Pesz, K.; Niklinski, J.; Chomczyk, M.A.; et al. DNA methylation abundantly associates with fetal alcohol spectrum disorder and its subphenotypes. Epigenomics 2019, 11, 767–785.
- Lussier, A.A.; Weinberg, J.; Kobor, M.S. Epigenetics studies of fetal alcohol spectrum disorder: Where are we now? Epigenomics 2017, 9, 291–311.
- Mews, P.; Egervari, G.; Nativio, R.; Sidoli, S.; Donahue, G.; Lombroso, S.I.; Alexander, D.C.; Riesche, S.L.; Heller, E.A.; Nestler, E.J.; et al. Alcohol metabolism contributes to brain histone acetylation. Nature 2019, 574, 717–721.
- Cravo, M.L.; Camilo, M.E. Hyperhomocysteinemia in chronic alcoholism: Relations to folic acid and vitamins B(6) and B(12) status. Nutrition 2000, 16, 296–302.
- Halsted, C.H.; Villanueva, J.A.; Devlin, A.M.; Niemelä, O.; Parkkila, S.; Garrow, T.A.; Wallock, L.M.; Shigenaga, M.K.; Melnyk, S.; James, S.J. Folate deficiency disturbs hepatic methionine metabolism and promotes liver injury in the ethanol-fed micropig. Proc. Natl. Acad. Sci. USA 2002, 99, 10072–10077.
- Muldoon, R.T.; McMartin, K.E. Ethanol acutely impairs the renal conservation of 5-methyltetrahydrofolate in the isolated perfused rat kidney. Alcohol. Clin. Exp. Res. 1994, 18, 333–339.
- Garro, A.J.; McBeth, D.L.; Lima, V.; Lieber, C.S. Ethanol consumption inhibits fetal DNA methylation in mice: Implications for the fetal alcohol syndrome. Alcohol. Clin. Exp. Res. 1991, 15, 395–398.
- Haycock, P.C.; Ramsay, M. Exposure of mouse embryos to ethanol during preimplantation development: Effect on DNA methylation in the h19 imprinting control region. Biol. Reprod. 2009, 81, 618–627.
- Liu, Y.; Balaraman, Y.; Wang, G.; Nephew, K.P.; Zhou, F.C. Alcohol exposure alters DNA methylation profiles in mouse embryos at early neurulation. Epigenetics 2009, 1, 500–511.
- Kaminen-Ahola, N.; Ahola, A.; Maga, M.; Mallitt, K.A.; Fahey, P.; Cox, T.C.; Whitelaw, E.; Chong, S. Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model. PLoS Genet. 2010, 15, e1000811.
- Wolff, G.L. Influence of maternal phenotype on metabolic differentiation of agouti locus mutants in the mouse. Genetics 1978, 88, 529–539.
- Dolinoy, D.C.; Weidman, J.R.; Waterland, R.A.; Jirtle, R.L. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ. Health Perspect. 2006, 114, 567–572.
- Waterland, R.A.; Jirtle, R.L. Transposable elements: Targets for early nutritional effects on epigenetic gene regulation. Mol. Cell. Biol. 2003, 23, 5293–5300.
- Marjonen, H.; Sierra, A.; Nyman, A.; Rogojin, V.; Gröhn, O.; Linden, A.M.; Hautaniemi, S.; Kaminen-Ahola, N. Early maternal alcohol consumption alters hippocampal DNA methylation, gene expression and volume in a mouse model. PLoS ONE 2015, 10, e0124931.
- Orkin, S.H.; Hochedlinger, K. Chromatin connections to pluripotency and cellular reprogramming. Cell 2011, 145, 835–850.
- Young, R.A. Control of the embryonic stem cell state. Cell 2011, 144, 940–954.
- Goodlett, C.R.; Horn, K.H. Mechanisms of alcohol-induced damage to the developing nervous system. Alcohol Res. Health 2001, 25, 175–184.
- Khalid, O.; Kim, J.J.; Kim, H.S.; Hoang, M.; Tu, T.G.; Elie, O.; Lee, C.; Vu, C.; Horvath, S.; Spigelman, I.; et al. Gene expression signatures affected by alcohol-induced DNA methylomic deregulation in human embryonic stem cells. Stem Cell Res. 2014, 12, 791–806.
- Sánchez-Alvarez, R.; Gayen, S.; Vadigepalli, R.; Anni, H. Ethanol diverts early neuronal differentiation trajectory of embryonic stem cells by disrupting the balance of lineage specifiers. PLoS ONE 2013, 8, e63794.
- Nash, R.; Krishnamoorthy, M.; Jenkins, A.; Csete, M. Human embryonic stem cell model of ethanol-mediated early developmental toxicity. Exp. Neurol. 2012, 234, 127–135.
- Taléns-Visconti, R.; Sanchez-Vera, I.; Kostic, J.; Perez-Arago, M.A.; Erceg, S.; Stojkovic, M.; Guerri, C. Neural differentiation from human embryonic stem cells as a tool to study early brain development and the neuroteratogenic effects of ethanol. Stem Cells Dev. 2011, 20, 327–339.
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75.
- Gao, W.; Zhou, P.; Ma, X.; Tschudy-Seney, B.; Chen, J.; Magner, N.L.; Revzin, A.; Nolta, J.A.; Zern, M.A.; Duan, Y. Ethanol negatively regulates hepatic differentiation of hESC by inhibition of the MAPK/ERK signaling pathway in vitro. PLoS ONE 2014, 9, e112698.
- Vangipuram, S.D.; Lyman, W.D. Ethanol affects differentiation-related pathways and suppresses Wnt signaling protein expression in human neural stem cells. Alcohol. Clin. Exp. Res. 2012, 36, 788–797.
- Wang, Q.; Song, J.W.; Liu, Y.; Zhao, X.X. Involvement of Wnt pathway in ethanol-induced inhibition of mouse embryonic stem cell differentiation. Alcohol 2017, 58, 13–18.
- Worley, S.L.; Vaughn, B.J.; Terry, A.I.; Gardiner, C.S.; DeKrey, G.K. Time- and dose-dependent effects of ethanol on mouse embryonic stem cells. Reprod. Toxicol. 2015, 57, 157–164.
- Chambers, I.; Tomlinson, S.R. The transcriptional foundation of pluripotency. Development 2009, 136, 2311–2322.
- Thomson, M.; Liu, S.J.; Zou, L.N.; Smith, Z.; Meissner, A.; Ramanathan, S. Pluripotency factors in embryonic stem cells regulate differentiation into germ layers. Cell 2011, 145, 875–889.
- Wang, Z.; Oron, E.; Nelson, B.; Razis, S.; Ivanova, N. Distinct lineage specification roles for NANOG, OCT4, and SOX2 in human embryonic stem cells. Cell Stem Cell 2012, 10, 440–454.
- Ying, L.; Mills, J.A.; French, D.L.; Gadue, P. OCT4 Coordinates with WNT Signaling to Pre-pattern Chromatin at the SOX17 Locus during Human ES Cell Differentiation into Definitive Endoderm. Stem Cell Rep. 2015, 5, 490–498.
- Ogony, J.W.; Malahias, E.; Vadigepalli, R.; Anni, H. Ethanol alters the balance of Sox2, Oct4, and Nanog expression in distinct subpopulations during differentiation of embryonic stem cells. Stem Cells Dev. 2013, 22, 2196–2210.
- Miller, M.W. Exposure to ethanol during gastrulation alters somatosensory-motor cortices and the underlying white matter in the macaque. Cereb. Cortex 2007, 17, 2961–2971.
- Waddington, C.H. The Strategy of the Genes; A Discussion of Some Aspects of Theoretical Biology; George Allen & Unwin: London, UK, 1957.
- Iovino, N.; Cavalli, G. Rolling ES cells down the Waddington landscape with Oct4 and Sox2. Cell 2011, 145, 815–817.
- Hicks, S.D.; Middleton, F.A.; Miller, M.W. Ethanol-induced methylation of cell cycle genes in neural stem cells. J. Neurochem. 2010, 114, 1767–1780.
- Zhou, F.C.; Balaraman, Y.; Teng, M.; Liu, Y.; Singh, R.P.; Nephew, K.P. Alcohol alters DNA methylation patterns and inhibits neural stem cell differentiation. Alcohol. Clin. Exp. Res. 2011, 35, 735–746.
- Liyanage, V.R.; Zachariah, R.M.; Davie, J.R.; Rastegar, M. Ethanol deregulates Mecp2/MeCP2 in differentiating neural stem cells via interplay between 5-methylcytosine and 5-hydroxymethylcytosine at the Mecp2 regulatory elements. Exp. Neurol. 2015, 265, 102–117.
- Veazey, K.J.; Carnahan, M.N.; Muller, D.; Miranda, R.C.; Golding, M.C. Alcohol-induced epigenetic alterations to developmentally crucial genes regulating neural stemness and differentiation. Alcohol. Clin. Exp. Res. 2013, 37, 1111–1122.
- Veazey, K.J.; Parnell, S.E.; Miranda, R.C.; Golding, M.C. Dose-dependent alcohol-induced alterations in chromatin structure persist beyond the window of exposure and correlate with fetal alcohol syndrome birth defects. Epigenetics Chromatin 2015, 8, 39.
- Wang, L.; Sun, H.; Pan, B.; Zhu, J.; Huang, G.; Huang, X.; Tian, J. Inhibition of histone acetylation by curcumin reduces alcohol-induced expression of heart development-related transcription factors in cardiac progenitor cells. Biochem. Biophys. Res. Commun. 2012, 424, 593–596.
- Zhong, L.; Zhu, J.; Lv, T.; Chen, G.; Sun, H.; Yang, X.; Huang, X.; Tian, J. Ethanol and its metabolites induce histone lysine 9 acetylation and an alteration of the expression of heart development-related genes in cardiac progenitor cells. Cardiovasc. Toxicol. 2010, 10, 268–274.
- Mandal, C.; Halder, D.; Jung, K.H.; Chai, Y.G. Maternal alcohol consumption and altered miRNAs in the developing fetus: Context and future perspectives. J. Appl. Toxicol. 2018, 38, 100–107.
- Sathyan, P.; Golden, H.B.; Miranda, R.C. Competing interactions between micro-RNAs determine neural progenitor survival and proliferation after ethanol exposure: Evidence from an ex vivo model of the fetal cerebral cortical neuroepithelium. J. Neurosci. 2007, 27, 8546–8557.
- Balaraman, S.; Winzer-Serhan, U.H.; Miranda, R.C. Opposing actions of ethanol and nicotine on microRNAs are mediated by nicotinic acetylcholine receptors in fetal cerebral cortical-derived neural progenitor cells. Alcohol. Clin. Exp. Res. 2012, 36, 1669–1677.
- Downing, C.; Johnson, T.E.; Larson, C.; Leakey, T.I.; Siegfried, R.N.; Rafferty, T.M.; Cooney, C.A. Subtle decreases in DNA methylation and gene expression at the mouse Igf2 locus following prenatal alcohol exposure: Effects of a methyl-supplemented diet. Alcohol 2011, 45, 65–71.
- Pan, B.; Zhu, J.; Lv, T.; Sun, H.; Huang, X.; Tian, J. Alcohol consumption during gestation causes histone3 lysine9 hyperacetylation and an alternation of expression of heart development-related genes in mice. Alcohol. Clin. Exp. Res. 2014, 38, 2396–2402.
- Peng, C.; Zhu, J.; Sun, H.C.; Huang, X.P.; Zhao, W.A.; Zheng, M.; Liu, L.J.; Tian, J. Inhibition of histone H3K9 acetylation by anacardic acid can correct the over-expression of Gata4 in the hearts of fetal mice exposed to alcohol during pregnancy. PLoS ONE 2014, 9, e104135.
- Peng, C.; Zhang, W.; Zhao, W.; Zhu, J.; Huang, X.; Tian, J. Alcohol-induced histone H3K9 hyperacetylation and cardiac hypertrophy are reversed by a histone acetylases inhibitor anacardic acid in developing murine hearts. Biochimie 2015, 113, 1–9.
- Zhang, W.; Peng, C.; Zheng, M.; Gao, W.; Zhu, J.; Lv, T.; Liu, L.; Liu, Z.; Li, H.; Xv, Y.; et al. Prenatal alcohol exposure causes the over-expression of DHAND and EHAND by increasing histone H3K14 acetylation in C57 BL/6 mice. Toxicol. Lett. 2014, 228, 140–146.
- Kaminen-Ahola, N.; Ahola, A.; Flatscher-Bader, T.; Wilkins, S.J.; Anderson, G.J.; Whitelaw, E.; Chong, S. Postnatal growth restriction and gene expression changes in a mouse model of fetal alcohol syndrome. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 818–826.
- Zhang, C.R.; Kurniawan, N.D.; Yamada, L.; Fleming, W.; Kaminen-Ahola, N.; Ahola, A.; Galloway, G.; Chong, S. Early gestational ethanol exposure in mice: Effects on brain structure, energy metabolism and adiposity in adult offspring. Alcohol 2019, 75, 1–10.
- Zhang, C.R.; Ho, M.F.; Vega, M.C.; Burne, T.H.; Chong, S. Prenatal ethanol exposure alters adult hippocampal VGLUT2 expression with concomitant changes in promoter DNA methylation, H3K4 trimethylation and miR-467b-5p levels. Epigenet. Chromatin 2015, 8, 40.
- Sanchez Vega, M.C.; Chong, S.; Burne, T.H. Early gestational exposure to moderate concentrations of ethanol alters adult behaviour in C57BL/6J mice. Behav. Brain Res. 2013, 252, 326–333.
More
Information
Subjects:
Genetics & Heredity
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
727
Revisions:
3 times
(View History)
Update Date:
03 Aug 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No