 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sai-Yang Zhang | + 1970 word(s) | 1970 | 2021-07-22 08:01:58 | | | |
| 2 | Amina Yu | Meta information modification | 1970 | 2021-08-03 06:10:22 | | |
Video Upload Options
FAK is an intracellular non-receptor tyrosine kinase that promotes tumor cell growth by controlling cell adhesion, migration, proliferation, and survival. Therefore, targeting FAK is considered to be a promising cancer therapy with small molecules.
1. Introduction
Receptor tyrosine kinases (RTK) and non-receptor tyrosine kinases play vital roles in extracellular signals and process of cancer cell migration, proliferation, and survival, which usually are overexpressed in many human cancer cells [1][2]. Therefore, targeting RTKs and non-receptor tyrosine kinases has been considered to be a promising therapy for the treatment of human cancers [3].
FAK is one of cytoplasmic non-receptor protein tyrosine kinase, which is a member of the focal adhesion complex family [4][5]. FAK is a 125 kDa protein that is recruited in the early stages of cell adhesion and phosphorylated to mediate the formation of local adhesion [6][7][8][9]. FAK gene exists on chromosome 8q24 and encodes 1052 amino acid residues and its protein structure consists of three parts: N -terminal region, kinase region, and C -terminal region [9].
FAK was first discovered in 1992, which is related to integrin signal transduction [4]. FAK is considered to be a key regulator of growth factor receptor and integrin-mediated signaling, which controls the basic processes of human normal or cancer cells through its kinase activity and scaffold function [10][11]. FAK overexpression is detected in various tumor types, including prostate cancer, breast cancer, lung cancer, ovarian cancer, and neck cancer, and FAK is also associated with the poor prognosis of cancer patients [12][13]. Besides, it is related to unregulated proteins in cancer, providing a perfect environment for FAK-specific treatment [14]. FAK regulates tumorigenic and metastatic potential through a highly coordinated signal network, thereby promoting the occurrence of malignant tumors. These signal networks coordinate a series of cellular processes, such as cell proliferation, survival, invasion, migration, angiogenesis, and the regulation of cancer stem cell activity [5][15][16]. Different FAK antisense oligonucleotides [17], dominant-negative FAK C -terminal domain, FAK-CD or FRNK or FAK-siRNA [18][19] can lead to decreased cell survival, growth inhibition, or apoptosis. In recent years, FAK has been considered as a new potential tumor-treatment target [20].
So far, some FAK inhibitors have been identified and proved to be effective inhibitors that inhibit the tumor growth and metastasis in several preclinical and clinical models [21][22][23][24]. TAE-226 is a typical FAK inhibitor (IC 50 = 5.5 nM), which has strong antiproliferative and anticancer potency against several types of cancers in vitro and in vivo [21]. GSK2256098 is a highly potent, ATP-competitive, and reversible FAK inhibitor [25], which is currently underway in clinical trials as a monotherapy or in combination with other drugs for patients with brain tumors or other cancers [26]. VS-6063 is a highly effective second-generation FAK inhibitor. Phase II clinical trials of VS-6063 in patients with KRAS mutant non-small cell lung cancer have been completed [27]. VS-6062 has now completed the first phase of research. VS-6062 not only exhibits significant antiproliferation potency of cancer cells but also plays a new role in changing tumor microenvironment [28]. VS-4718 significantly affects cell viability by inhibiting FAK activity and locking Y397 phosphorylation [23][24]. Another novel FAK inhibitor, CEP-37440 , is currently in clinical trials for the treatment of cancers [22] ( Table 1 ). The proteolytic targeting chimera (PROTAC) that can induce FAK degradation by using FAK kinase inhibitors as a “warhead” also has been developed [29]. A large number of studies have provided convincing evidence that FAK inhibitors could be used as chemical-targeted drugs to induce growth inhibition and apoptosis of cancer cells [30][31][32]. This article briefly introduces the structure and biological functions of FAK. Aiming at the development of FAK drugs, it focuses on the design, chemical types, and activities of FAK drugs to provide references for the development of new antitumor drugs.
2. FAK Structure and Function
The FAK protein molecule can be roughly divided into N -terminal FERM region, middle kinase region, C -terminal focal adhesion target (FAT), and proline-rich regions (PRR1, PRR2, and PRR3). FAK has 6 tyrosine sites that can be phosphorylated: Y397, Y407, Y576, Y577, Y861, and Y925, all of them are the key parts in FAK’s signal transduction. Y397 and Y407 sites are located at the N -terminus, Y576 and Y577 are located in the activation loop of the kinase region, and Y861 and Y925 are located at the C -terminus. Y397 is an autophosphorylation site of FAK, which provides a high-affinity-binding site for Src family proteins and plays a vital role in downstream signaling pathways [16].
There are binding sites for a variety of proteins at the N -terminus, such as signal transduction proteins, cytoskeleton proteins, and integrin β subunits [33]. The N -terminal FERM domain is composed of three closely related subdomains F1, F2, and F3, forming a three-leaf clover-like structure [34] ( Figure 1 ). Among of them, F1 and F2 subdomains can interact with p53 and hinder the apoptosis of tumor cells; the F2 subdomain can regulate kinase-independent activities and mediate cell survival; the F3 subdomain can interact with Mdm-2 to enhance ubiquitination of p53 [33]. The kinase domain of FAK also called the catalytic domain, has a highly conserved amino acid sequence that can phosphorylate corresponding amino acid residues in proteins such as PI3K, growth factor receptor-binding protein 2 (GRB2), Src, and Cas. The focal adhesion targeting region at the C -terminus could interact with focal adhesion proteins, including paxillin [35], Talin [36], and Vinculin [37]. FAT can also bind to the F2 leaf in the FERM domain, leading to the dimerization of FAK, which is of great significance to the activation and nuclear localization of FAK [38]. PRR1 is located between the FERM region and the kinase region and has a binding site to the SH3 domain, which can bind to proteins such as Src. PRR2 and PRR3 near the C -terminus can mediate the interaction between FAK and protein molecules containing SH3 domains.
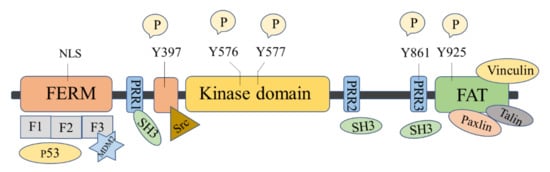
FAK shows kinase-dependent enzyme function and a kinase-independent scaffold function, which play important roles in regulating the survival, proliferation, migration, and angiogenesis of cancer cells. FAK is overexpressed in many human cancers, including glioma, breast, colon, prostate, and other cancers [10][16][39]. Inhibition of FAK has been shown to inhibit tumor progression and metastasis.
3. The Discovery of FAK Inhibitors and Their Therapeutic Significance
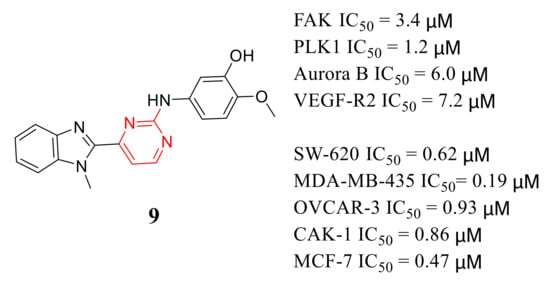
2-Anilino-4-(benzimidazol-2-yl ) pyrimidine 9 is a multi-kinase inhibitor with inhibitory potency of FAK, PLK1, Aurora B, and VEGF-R2 with IC 50 values of 3.4, 1.2, 6, and 7.2 μM, respectively ( Figure 2 ). Compound 9 also exhibited potent inhibition growth of SW-620, MDA-MB-435, OVCAR-3, CAKI-1, and MCF-7 cells at micromolar concentrations (IC 50 values = 0.62, 0.19, 0.93, 0.86 and 0.47 μM, respectively) [40].
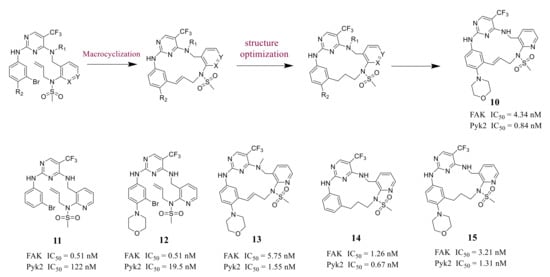
Farand’s group reported novel macrocycle diphenylpyrimidine derivatives as FAK inhibitors based on the lead compound VS-6062 in 2016 ( Figure 3 ). The representative compound 10 showed strong antagonism against FAK and Pyk2 with IC 50 values of 4.34 nM and 0.84 nM, respectively [41]. Among these compounds, compound 10 showed the best metabolic stability in the analysis of human microsomes.
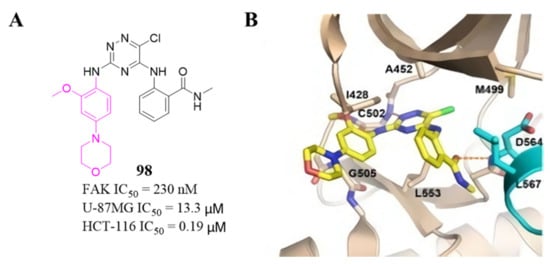
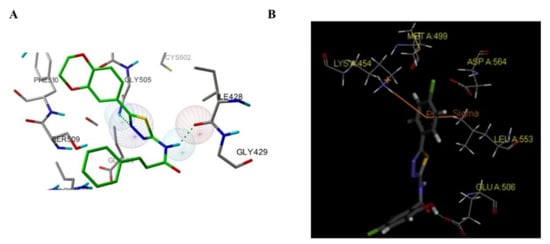
Among these compounds, compound 98 containing a 2- OCH 3-4-morpholino group showed the best anti-FAK activity with an IC 50 value of 230 nM and excellent antiproliferation activity against HCT-116 cells (IC 50 = 0.19 µM). The results of the docking studies suggested that the van der Waals interactions between the chlorine atom and Met499 of FAK protein might not be important to the binding of compound 98 with FAK protein, suggesting that the weaker hydrogen bonds between the amino 1,2,4-triazine and the hinge region of FAK may play an important role in the inhibitory activity of compound 98 ( Figure 4 ).
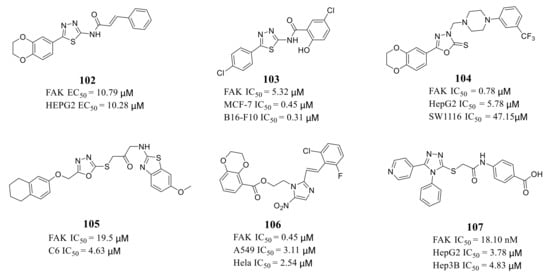
Zhu’s group reported a new class of novel 1,3,4-thiadiazoles as FAK inhibitors. The most active compound 102 inhibited the activity of FAK and HEPG2 cells with an EC 50 value of 10.79 and 10.28 μM, respectively ( Figure 5 ). Furthermore, Zhu’s group designed and synthesized 1,3,4-thiadiazol-2-amide derivatives with more potent inhibitory potency of FAK and cancer cells growth compared to 102 [42]. The most active compound 103 exhibited better inhibitory activity against FAK with an IC 50 value of 5.32 μM and inhibited proliferation of MCF-7 and B16-F10 cells with IC 50 values of 0.45 and 0.31 μM, respectively [43]. The results of docking studies suggested that compounds 102 and 103 could be well embedded in the ATP-binding pocket of FAK through a hydrogen bond, π–σ interaction, and π–cation interaction ( Figure 6 ).
4. The Discovery of FAK-Targeting PROTACs and Their Therapeutic Significance
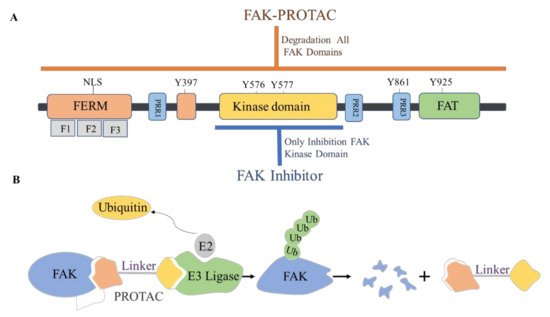
The proteolytic targeting chimera (PROTAC) is a new chemical knockout technique for post-translational proteins. PROTAC is a heterogeneous bifunctional small molecule containing two recognition parts: one specifically binds to E3 ubiquitin ligase, and the other specifically binds to the target protein. The PROTAC molecule can drive the E3 ubiquitin ligase to the target protein, which leads to the ubiquitination of the target protein, which leads to proteasome-mediated degradation ( Figure 7 ) [44][45].
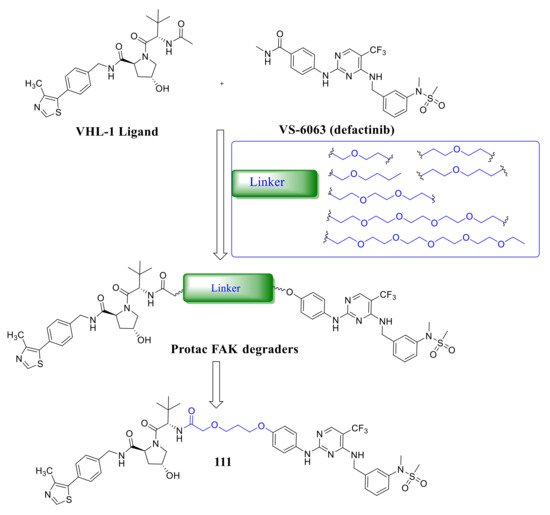
Crew’s group reported potent FAK selective degraders in 2018 [46]. In this work, they designed FAK degraders by conjugating a FAK ligand derived from VS-6063 ( defactinib ) through slight modifications with a peptide-based VHL ligand or thalidomide through ether linkers ( Figure 8 ). All of these FAK PROTACs exhibited good inhibitory potency against FAK and could significantly induce the degradation of FAK at low concentrations. Among the FAK-PROTACs, compound 111 exhibited potent activity, which showed an IC 50 value of 6.5 nM, DC 50 value of 3 nM, and Dmax over 99%. Compound 111 displayed better selectivity to FAK than VS-6063 . In addition, compound 111 displayed significant effects in suppressing the FAK downstream signaling pathways, which could highly induce the degradation of FAK in a dose-dependent manner with only 34% total FAK remaining at 10 nM and 5% at 50 nM, which outperformed VS-6063 ( defactinib ). In addition, compound 111 significantly reduces p -paxillin levels by as much as 85–90% at 50 nM, p -Akt suppression of 93% at 1 μM, and is more effective than VS-6063 . Compound 111 also significantly inhibited the migration and invasion of MDA-MB-23 cells.
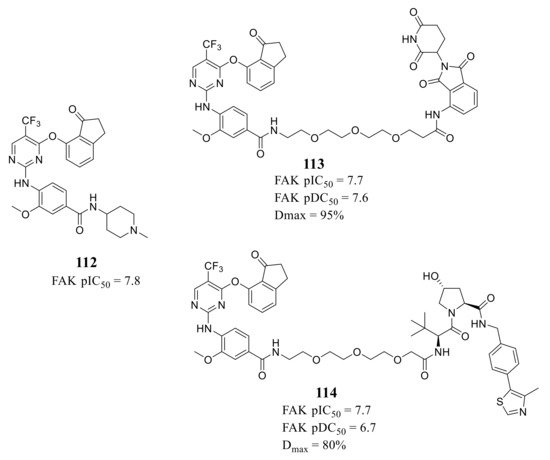
Ettmayer’s group reported a new class of FAK-PROTACs represented by compounds 113 and 114 in 2019, by tethering a previously identified FAK inhibitor compound 112 through an ether linker with a peptide-based VHL ligand and pomalidomide, respectively [47] ( Figure 9 ). Both of FAK-PROTACs (compound 113 and 114) showed comparable degradation potencies and efficacies with mean pDC 50 values of 7.45 and 7.08 nM, respectively. The CRBN-based PROTAC 113 potently degraded FAK (DC 50 = 27 nM) with a Dmax of 95% at the chosen experimental conditions, while the VHL-based PROTAC 114 only achieved a Dmax of 80% and was less potent (DC 50 = 243 nM) in the A549 cells. However, although the potent depletion of FAK, both FAK-PROTACs (compound 113 and 114) did not exhibit antiproliferation activity against the tested cell lines in either short or long-term assays, which indicated that scaffolding function of FAK may not be required for the proliferation of the tested cell lines.
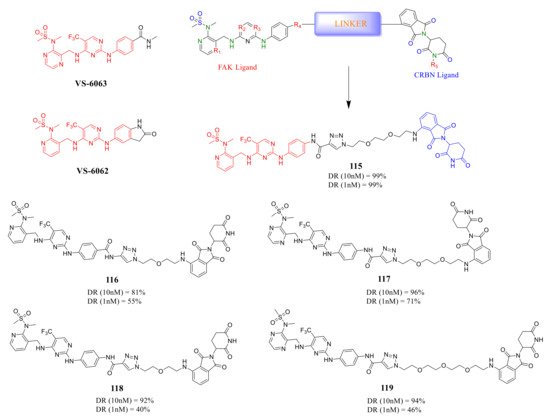
Rao’s group reported a new class of FAK-PROTACs in 2020, by tethering a previously identified FAK inhibitors VS-6062 or VS-6063 through different ether linker with a CRBN E3 ligand pomalidomide [48] ( Figure 10 ). They noticed that shorter diethylene or triethylene glycol linkers exhibited higher degradation activity of FAK. Among them, compound 115 caused rapid and significant degradation of FAK with DC 50 values at the scale of picomolar in TM3, PA1, MDA-MB-436, LNCaP, and Ramos cells (DC 50 = 310, 80, 330, 370, and 40 pM, respectively). Compound 115 showed a significant inhibitory effect on the autophosphorylation of FAK (pFAK tyr397 ) below 1 nM, but VS-6062 showed an inhibitory effect on pFAK tyr397 only at 3 μM. However, the efficient knockdown of FAK by compound 115 did not more severely affect the proliferation of the tested cell lines.
References
- Cui, J.J. A new challenging and promising era of tyrosine kinase inhibitors. ACS Med. Chem. Lett. 2014, 5, 272–274.
- Kong, X.; Pan, P.; Sun, H.; Xia, H.; Wang, X.; Li, Y.; Hou, T. Drug Discovery Targeting Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2019, 62, 10927–10954.
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934.
- Schaller, M.D.; Borgman, C.A.; Cobb, B.S.; Vines, R.R.; Reynolds, A.B.; Parsons, J.T. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc. Natl. Acad. Sci. USA 1992, 89, 5192–5196.
- Zhao, J.; Guan, J.-L. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009, 28, 35–49.
- Schaller, M.D. The focal adhesion kinase. J. Endocrinol. 1996, 150, 1–7.
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005, 6, 56–68.
- Parsons, J.T. Focal adhesion kinase: The first ten years. J. Cell Sci. 2003, 116, 1409–1416.
- Hildebrand, J.D.; Schaller, M.D.; Parsons, J.T. Identification of sequences required for the efficient localization of the Focal Adhesion Kinase, pp125(FAK), to cellular focal adhesions. J. Cell Biol. 1993, 123, 993–1005.
- Schaller, M.D. Cellular functions of FAK kinases: Insight into molecular mechanisms and novel functions. J. Cell Sci. 2010, 123, 1007–1013.
- Frame, M.C.; Patel, H.; Serrels, B.; Lietha, D.; Eck, M.J. The FERM domain: Organizing the structure and function of FAK. Nat. Rev. Mol. Cell Biol. 2010, 11, 802–814.
- Béraud, C.; Dormoy, V.; Danilin, S.; Lindner, V.; Béthry, A.; Hochane, M.; Coquard, C.; Barthelmebs, M.; Jacqmin, D.; Lang, H.; et al. Targeting FAK scaffold functions inhibits human renal cell carcinoma growth. Int. J. Cancer 2015, 137, 1549–1559.
- Pylayeva, Y.; Gillen, K.M.; Gerald, W.; Beggs, H.E.; Reichardt, L.F.; Giancotti, F.G. Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J. Clin. Investig. 2009, 119, 252–266.
- Siesser, P.M.F.; Hanks, S.K. The signaling and biological implications of FAK overexpression in cancer. Clin. Cancer Res. 2006, 12, 3233–3237.
- Lee, B.Y.; Timpson, P.; Horvath, L.G.; Daly, R.J. FAK signaling in human cancer as a target for therapeutics. Pharmacol. Ther. 2015, 146, 132–149.
- Lechertier, T.; Hodivala-Dilke, K. Focal Adhesion Kinase and tumour angiogenesis. J. Pathol. 2012, 226, 404–412.
- Smith, C.S.; Golubovskaya, V.M.; Peck, E.; Xu, L.-H.; Monia, B.P.; Yang, X.; Cance, W.G. Effect of focal adhesion kinase (FAK) downregulation with FAK antisense oligonucleotides and 5-fluorouracil on the viability of melanoma cell lines. Melanoma Res. 2005, 15, 357–362.
- Halder, J.; Landen, C.N., Jr.; Lutgendorf, S.K.; Li, Y.; Jennings, N.B.; Fan, D.; Nelkin, G.M.; Schmandt, R.; Schaller, M.D.; Sood, A.K. Focal adhesion kinase silencing augments docetaxel-mediated apoptosis in ovarian cancer cells. Clin. Cancer Res. 2005, 11, 8829–8836.
- Demirbas, A.; Sahin, D.; Demirbas, N.; Karaoglu, S.A. Synthesis of some new 1,3,4-thiadiazol-2-ylmethyl-1,2,4-triazole derivatives and investigation of their antimicrobial activities. Eur. J. Med. Chem. 2009, 44, 2896–2903.
- Kuhn, M.W.; Armstrong, S.A. Designed to kill: Novel menin-MLL inhibitors target MLL-rearranged leukemia. Cancer Cell 2015, 27, 431–433.
- Shi, Q.; Hjelmeland, A.B.; Keir, S.T.; Song, L.; Wickman, S.; Jackson, D.; Ohmori, O.; Bigner, D.D.; Friedman, H.S.; Rich, J.N. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Mol. Carcinog. 2007, 46, 488–496.
- Ott, G.R.; Cheng, M.; Learn, K.S.; Wagner, J.; Gingrich, D.E.; Lisko, J.G.; Curry, M.; Mesaros, E.F.; Ghose, A.K.; Quail, M.R.; et al. Discovery of Clinical Candidate CEP-37440, a Selective Inhibitor of Focal Adhesion Kinase (FAK) and Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2016, 59, 7478–7496.
- Shimizu, T.; Fukuoka, K.; Takeda, M.; Iwasa, T.; Yoshida, T.; Horobin, J.; Keegan, M.; Vaickus, L.; Chavan, A.; Padval, M.; et al. A first-in-Asian phase 1 study to evaluate safety, pharmacokinetics and clinical activity of VS-6063, a focal adhesion kinase (FAK) inhibitor in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2016, 77, 997–1003.
- Kang, Y.; Hu, W.; Ivan, C.; Dalton, H.J.; Miyake, T.; Pecot, C.V.; Zand, B.; Liu, T.; Huang, J.; Jennings, N.B.; et al. Role of Focal Adhesion Kinase in Regulating YB–1–Mediated Paclitaxel Resistance in Ovarian Cancer. JNCI J. Natl. Cancer Inst. 2013, 105, 1485–1495.
- Brown, N.F.; Williams, M.; Arkenau, H.T.; Fleming, R.A.; Tolson, J.; Yan, L.; Zhang, J.; Singh, R.; Auger, K.R.; Lenox, L.; et al. A study of the focal adhesion kinase inhibitor GSK2256098 in patients with recurrent glioblastoma with evaluation of tumor penetration of [11C]GSK2256098. Neuro Oncol. 2018, 20, 1634–1642.
- Mak, G.; Soria, J.C.; Blagden, S.P.; Plummer, R.; Fleming, R.A.; Nebot, N.; Zhang, J.; Mazumdar, J.; Rogan, D.; Gazzah, A.; et al. A phase Ib dose-finding, pharmacokinetic study of the focal adhesion kinase inhibitor GSK2256098 and trametinib in patients with advanced solid tumours. Br. J. Cancer 2019, 120, 975–981.
- Jones, S.F.; Siu, L.L.; Bendell, J.C.; Cleary, J.M.; Razak, A.R.A.; Infante, J.R.; Pandya, S.S.; Bedard, P.L.; Pierce, K.J.; Houk, B.; et al. A phase I study of VS-6063, a second-generation focal adhesion kinase inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 1100–1107.
- Stokes, J.B.; Adair, S.J.; Slack-Davis, J.K.; Walters, D.M.; Tilghman, R.W.; Hershey, E.D.; Lowrey, B.; Thomas, K.S.; Bouton, A.H.; Hwang, R.F.; et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol. Cancer Ther. 2011, 10, 2135–2145.
- Lu, Y.; Sun, H. Progress in the Development of Small Molecular Inhibitors of Focal Adhesion Kinase (FAK). J. Med. Chem. 2020, 63, 14382–14403.
- Fu, Q.; Socki, R.A.; Niles, P.B. Evaluating reaction pathways of hydrothermal abiotic organic synthesis at elevated temperatures and pressures using carbon isotopes. Geochim. Cosmochim. Acta 2015, 154, 1–17.
- Rosado, P.; Lequerica-Fernández, P.; Peña, I.; Alonso-Durán, L.; de Vicente, J.C. In oral squamous cell carcinoma, high FAK expression is correlated with low P53 expression. Virchows Arch. 2012, 461, 163–168.
- Zheng, Y.; Gierut, J.; Wang, Z.; Miao, J.; Asara, J.M.; Tyner, A.L. Protein tyrosine kinase 6 protects cells from anoikis by directly phosphorylating focal adhesion kinase and activating AKT. Oncogene 2013, 32, 4304–4312.
- Lim, S.-T.; Chen, X.L.; Lim, Y.; Hanson, D.A.; Vo, T.-T.; Howerton, K.; Larocque, N.; Fisher, S.J.; Schlaepfer, D.D.; Ilic, D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol. Cell 2008, 29, 9–22.
- Hamadi, A.; Bouali, M.; Dontenwill, M.; Stoeckel, H.; Takeda, K.; Rondé, P. Regulation of focal adhesion dynamics and disassembly by phosphorylation of FAK at tyrosine 397. J. Cell Sci. 2005, 118, 4415–4425.
- Scheswohl, D.M.; Harrell, J.R.; Rajfur, Z.; Gao, G.; Campbell, S.L.; Schaller, M.D. Multiple paxillin binding sites regulate FAK function. J. Mol. Signal. 2008, 3, 1.
- Lawson, C.; Lim, S.-T.; Uryu, S.; Chen, X.L.; Calderwood, D.A.; Schlaepfer, D.D. FAK promotes recruitment of talin to nascent adhesions to control cell motility. J. Cell Biol. 2012, 196, 223–232.
- Subauste, M.C.; Pertz, O.; Adamson, E.D.; Turner, C.E.; Junger, S.; Hahn, K.M. Vinculin modulation of paxillin–FAK interactions regulates ERK to control survival and motility. J. Cell Biol. 2004, 165, 371–381.
- Cho, J.-H.; Muralidharan, V.; Vila-Perello, M.; Raleigh, D.P.; Muir, T.W.; Palmer, A.G. Tuning protein autoinhibition by domain destabilization. Nat. Struct. Mol. Biol. 2011, 18, 550–555.
- Golubovskaya, V.M.; Kwen, F.A.; Cance, W.G. Focal adhesion kinase and cancer. Histol. Histopathol. 2009, 24, 503–510.
- Determann, R.; Dreher, J.; Baumann, K.; Preu, L.; Jones, P.G.; Totzke, F.; Schachtele, C.; Kubbutat, M.H.; Kunick, C. 2-Anilino-4-(benzimidazol-2-yl)pyrimidines-a multikinase inhibitor scaffold with antiproliferative activity toward cancer cell lines. Eur. J. Med. Chem. 2012, 53, 254–263.
- Farand, J.; Mai, N.; Chandrasekhar, J.; Newby, Z.E.; van Veldhuizen, J.; Loyer-Drew, J.; Venkataramani, C.; Guerrero, J.; Kwok, A.; Li, N.; et al. Selectivity switch between FAK and Pyk2: Macrocyclization of FAK inhibitors improves Pyk2 potency. Bioorg. Med. Chem. Lett. 2016, 26, 5926–5930.
- Sun, J.; Yang, Y.-S.; Li, W.; Zhang, Y.-B.; Wang, X.-L.; Tang, J.-F.; Zhu, H.-L. Synthesis, biological evaluation and molecular docking studies of 1,3,4-thiadiazole derivatives containing 1,4-benzodioxan as potential antitumor agents. Bioorg. Med. Chem. Lett. 2011, 21, 6116–6121.
- Yang, X.-H.; Xiang, L.; Li, X.; Zhao, T.-T.; Zhang, H.; Zhou, W.-P.; Wang, X.-M.; Gong, H.-B.; Zhu, H.-L. Synthesis, biological evaluation, and molecular docking studies of 1,3,4-thiadiazol-2-amide derivatives as novel anticancer agents. Bioorg. Med. Chem. 2012, 20, 2789–2795.
- Sun, Y.; Zhao, X.; Ding, N.; Gao, H.; Wu, Y.; Yang, Y.; Zhao, M.; Hwang, J.; Song, Y.; Liu, W.; et al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 2018, 28, 779–781.
- Raina, K.; Crews, C.M. Chemical Inducers of Targeted Protein Degradation*. J. Biol. Chem. 2010, 285, 11057–11060.
- Kargbo, R.B. Bifunctional Pyrimidines as Modulators of Focal Adhesion Kinase. ACS Med. Chem. Lett. 2020, 11, 409–411.
- Popow, J.; Arnhof, H.; Bader, G.; Berger, H.; Ciulli, A.; Covini, D.; Dank, C.; Gmaschitz, T.; Greb, P.; Karolyi-Özguer, J.; et al. Highly Selective PTK2 Proteolysis Targeting Chimeras to Probe Focal Adhesion Kinase Scaffolding Functions. J. Med. Chem. 2019, 62, 2508–2520.
- Gao, H.; Wu, Y.; Sun, Y.; Yang, Y.; Zhou, G.; Rao, Y. Design, Synthesis, and Evaluation of Highly Potent FAK-Targeting PROTACs. ACS Med. Chem. Lett. 2020, 11, 1855–1862.


