1000/1000
Hot
Most Recent
 +1 point
+1 point
Mycobacterium tuberculosis (Mtb) is the microorganism that causes tuberculosis. The discovery of the antituberculosis agents in the 20th century has managed to improve the recovery rate and reduce the death rate tremendously. However, the conventional antituberculosis therapy is complicated by the development of resistant strains and adverse drug reactions experienced by the patients. Targeted drug delivery may be a potentially useful approach to be developed into clinically accepted treatment modalities. Active targeting utilizes a specifically designed targeting agent to deliver a chemically conjugated drug(s) towards Mtb. Passive targeting is very widely explored, with the development of multiple types of nanoparticles from organic and inorganic materials. The nanoparticles will be engulfed by macrophages and this will eliminate the Mtb that is present in the macrophages, or the encapsulated drug may be released at the sites of infections that may be in the form of intra- and extrapulmonary tuberculosis.
Before the discovery of current therapeutic agents, different treatment approaches such as herbal remedies, surgical intervention, and climate change were proposed to ease the lung consumption experienced by TB patients. However, most of these interventions were unsuccessful, leading to a high mortality rate among those infected.
In the 1940s, the discovery of penicillin as the first anti-infective initiated the discovery of many other anti-infective agents in the world. The discovery of rifampicin in the 1970’s managed to halve the duration of treatment from 18 months to 9 months, and with the subsequent addition of pyrazinamide, the minimum effective duration of treatment was further shortened to 6 months. The treatment options have expanded since then, with more drugs added into the regimen, providing options even in difficult cases such as resistant TB. Figure 1summarizes the medications and treatment plans against TB. First-line antituberculosis drugs known as isoniazid (INH/H), rifampicin (RIF/R), pyrazinamide (PZA/Z), and ethambutol (EMB/E) (Figure 2) were established and extensively used for more than half a century [1]. Patients would need to undergo six months of treatment under two cycles of chemotherapy; the intensive phase with a HRZE regimen for two months, followed by 4 months of the continuation phase under a HR regimen [2].
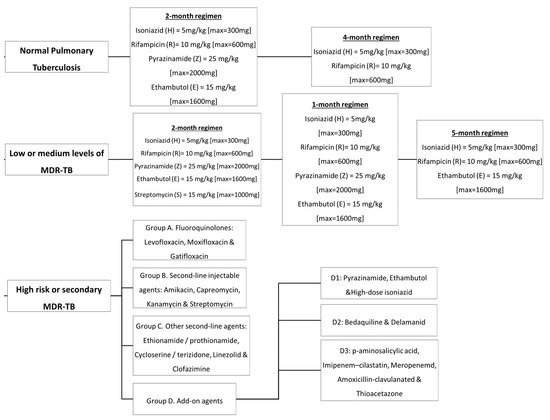
Figure 1. Antituberculosis medications and treatment plans available to date.
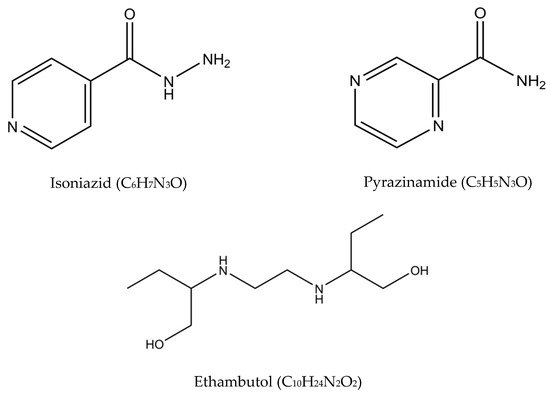
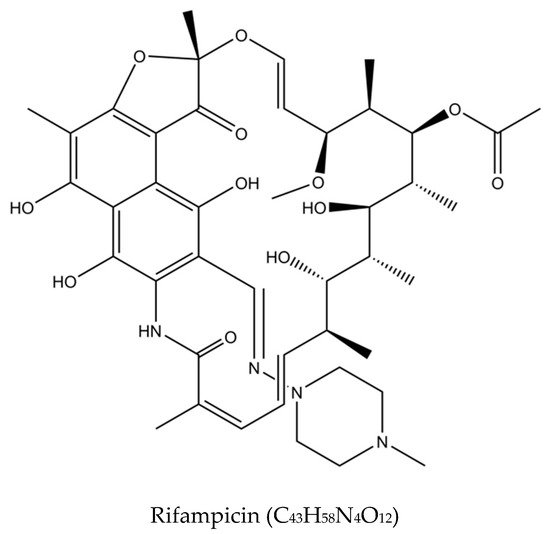
Figure 2. Molecular structures of the isoniazid, rifampicin, pyrazinamide, and ethambutol.
The intensive phase reduces the bacillary load by killing about 95% of the microorganisms, while the continuation phase focuses on eliminating the drug-sensitive organisms [3]. This treatment regimen successfully achieves an 85% success rate for new TB cases as reported by the WHO [4]. Despite the high success rate, HRZE contributes to several adverse events, including hepatotoxicity, hyperuricemia, peripheral neuritis, hypersensitivity, visual toxicity, cutaneous reactions, flu-like syndrome, ototoxicity, fever, gastrointestinal intolerance, and psychiatric changes. The prevalence of adverse effects is more significant in elderly patients as the susceptibility increases with age [5][6][7]. Table 1outlines the details on the first-line drugs with their targets, mechanism of actions, and mutations that may occur in Mtb that render them ineffective.
Table 1. Detailed description of first-line drugs with the target actions and gene mutations that may cause drug-resistance. Adopted from [4][8].
| Drug | Target | Target Actions to Mtb | Gene Mutations |
|---|---|---|---|
| Isoniazid (INH) | InhA [Enoyl-(acyl carrier-protein) reductase] |
Bactericidal: Cell envelope disruption. INH inhibits mycolic acid biosynthesis, an essential component of Mtb cell envelope. It specifically inhibits InhA, the enoyl reductase of Mtb, by forming a covalent adduct with the NAD cofactor. The INH/NAD adduct acts as a slow, tight-binding competitive inhibitor of InhA. | Mutation in genes katG, AhpC and inhA causing resistance to INH. |
| Rifampicin (RIF) | Bacterial RNA polymerase (RNAP) | Bactericidal: Inhibits Transcription RIF inhibits bacterial DNA-dependent RNA polymerase by forming a stable enzyme-drug complex with the β-subunit of RNA polymerase (RNAP-Rif), rpoB gene. It has a broad antibacterial spectrum, including activity against several forms of Mycobacterium. |
Mutation in codon 426-452 in the rpoB gene occurs at specific 81-base pair hot spot region causing RIF resistance. |
| Pyrazinamide (PZA) | S1 component of 30S Ribosomal subunit |
Bactericidal: Acidifies cytoplasm PZA inhibits translation and trans-translation. The active moiety of pyrazinamide is pyrazinoic acid (POA). POA is thought to disrupt membrane energy and inhibit membrane transport function at acidic pH. Its analogs have been shown to inhibit the activity of purified FAS I. | Mutation in the pncA gene and the changes at 561 bp and 82 bp causing PZA resistance. |
| Ethambutol (EMB) | Inhibits arbinosyltransferase |
Bacteriostatic: Cell wall disruption. EMB disrupts arabinogalactan synthesis, thereby preventing the interaction of 5’-hydroxyl groups of D-arabinose residues of arabinogalactan with mycolic acids that form a mycolyl-arabinogalactan-peptidoglycan complex of the Mtb cell wall. | Mutation of the EMB gene by protein-altering structure or by self-over expression will cause loss of EMB efficiency. Mutation in the gene embB at position 306 may occur by replacement of a single methionine with leucine or isoleucine, resulting in EMB resistance. |
Besides adverse effects, first-line antituberculosis treatment is also unsuitable for patients infected with drug-resistant Mtb strains. Drug resistance can develop due to several factors, including patients’ non-compliance towards the long treatment regimen, poor patient adherence to medication, and an inadequate and inappropriate treatment regimen by health providers [9][10]. This leads to the transmission of resistant bacilli to healthy individuals and, subsequently, the development of drug resistance [11]. In addition, the Mtb itself may develop a sophisticated natural defense mechanism to sustain life that will lead to drug resistance [12]. Gene mutations of Mtb may also play roles in which the encoding of drug targets or drug-activating enzymes could be altered [13].
Multidrug-resistant tuberculosis (MDR-TB) is said to occur when Mtb is resistant to at least two drugs: isoniazid and rifampicin [14]. MDR-TB occurrence causes more significant impact on controlling the transmission since isoniazid and rifampicin played an essential role in the TB management approach. The treatment options available for MDR-TB are expensive and require at least 18 months of therapy [8]. In 2016, the WHO established a guideline to a shorten the MDR-TB treatment regimen of about 9–12 months as the first option in patients with MDR-TB or rifampicin-resistant TB (RR-TB) that are not resistant to fluoroquinolones or second-line injectable agents. The drugs of choice are as follows: (A) second-line drugs (fluoroquinolones-levofloxacin, moxifloxacin and gatifloxacin), (B) second-line injectable drugs (amikacin, kanamycin, capreomycin and streptomycin), (C) second-line agents such as ethionamide, prothionamide, cycloserine, and linezolid, and (D) agents such as para-amino salicylic acid, pyrazinamide, ethambutol, high doses of isoniazid, bedaquiline, and delamanid (Figure 3) [15]. Although these drugs effectively treat MDR-TB and RR-TB, the genetic mutation of Mtb may still occur and produce resistance towards these drugs and hinder the TB treatment efficacy. The details of the second-line drugs’ targets and possible gene mutations are listed in Table 2.
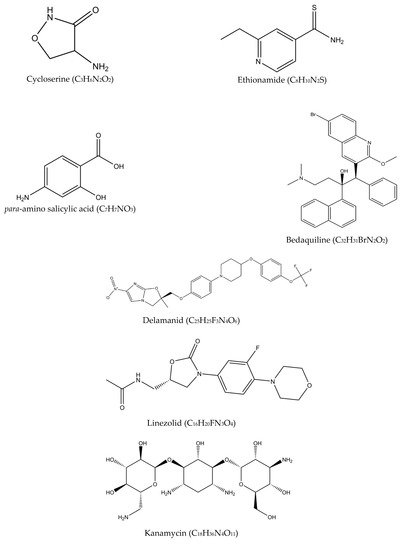
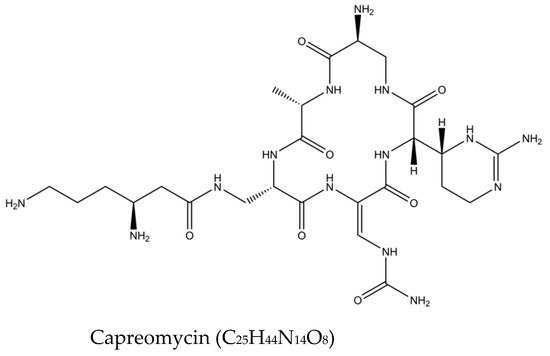
Figure 3. Molecular structures of the second-line drugs.
Table 2. Detailed description of second-line drugs with the target actions and gene mutations that may cause drug resistance. Adopted from [8].
| Drug | Target Actions to Mtb | Gene Mutations |
|---|---|---|
| Fluoroquinolones | Acts by interfering with mycobacterial DNA replication and transcription by inhibiting the topoisomerase II (DNA gyrase) enzyme, a tetramer with β and α subunit. | Chromosomal mutation of the genes gyrA and gyrB encoding DNA gyrase at position 90 and 94. |
| Kanamycin, capreomycin, amikacin, viomycin |
Alter the 16S rRNA level by interfering with the protein synthesis. | Mutation in the tylA gene causes resistance to capreomycin and viomycin. Mutation in 23S rRNA of the gene rrs also results in capreomycin and viomycin resistance. |
| Ethionamide | Inhibits the NADH-dependent ACP reductase enzyme by interfering with the mycolic acid biosynthesis that forms adduct with NAD. | Mutation in gene ethA, ethR, and inhA causing ethionamide resistance. |
| Cycloserine | Analog to alanine and acts by interfering with the biosynthesis of peptidoglycan in the Mycobacterial cell wall. | Overexpression of D-alanine racemose encoded by the gene alrA caused cycloserine resistance. |
| Linezolid | Binds to the 50S ribosomal subunit and inhibits early synthesis of protein. | Mutation in 23S rRNA also induces resistance to linezolid by interfering in the drug binding sites. |
| para-amino salicylic acid | Analog to aminobenzoic acid and interferes with the folate synthesis process. | Missense mutation in this folC gene causes resistance to para-amino salicylic acid. |
| Bedaquiline | Degrades the cell membrane of the Mycobacterium and interferes with ATP synthase encoded by the AtpE gene. | Mutation in the AtpE gene (A63 P,166 M) that codes for the subunit C of the Fo complex, inhibiting the ATP synthesis. |
| Delamanid | Active against non-growing persistent bacilli and acts by turning on the enzyme F420-dependent nitro reductase encoded by the ddn gene. | Mutations in the fgd1 and ddn genes give rise to resistant strains. |
To assess the presence of MDR-TB and RR-TB, the WHO guideline emphasizes bacteriological confirmation of TB and drug-resistant testing that involves rapid molecular tests, culture methods, or sequencing technologies. The corresponding treatment should take at least 9 months and up to 20 months using second-line drugs with counselling and monitoring of adverse events [16].
Due to the development of resistance, the search for new and effective antituberculosis agents has been continuously conducted with the hope to simplify (oral-only regimen) and shorten the treatment length (six months), while ensuring the safety and reducing the toxicity associated with current treatment options. There are also efforts for repurposing the use of other drugs as antituberculosis agents; among these are linezolid (oxazolidinone) and kanamycin (aminoglycosides), which have been shown to have significant activity on the mycobacteria [17].
Based on the list of second-line drugs tabulated in Table 2, only two drugs were newly approved specifically as antituberculosis drugs in the last 50 years—bedaquiline and delamanid. The two drugs are recommended by the WHO in the treatment of resistant TB under certain conditions, in combination with other conventional antituberculosis medications. Although research is continuously conducted worldwide, the discovery is rather challenging, lengthy, and costly. As antituberculosis drugs are generally used in combination regimens, testing for new combinations requires detailed information on the respective drugs’ safety, toxicity, and potential drug-drug interactions, in addition to their activity in special disease populations such as HIV/AIDS patients. Developing a new treatment combination with three or four new drugs, on the other hand, may require 20 to 30 years if it is performed one drug at a time [18]. These are among the challenges faced in the discovery and development of new antituberculosis drugs.
Another approach that is actively being investigated is the development of targeted treatments. This approach uses currently available antituberculosis drugs and improves the delivery through either an active or passive targeting. A multitude of research has been carried out over the past decades to explore the potential of this approach in TB treatment.
The concept of drug targeting aims to increase the accumulation of an active pharmaceutical ingredient (API) in a specific region of the body. The API, or the drug molecule, should be able to effectively cross the biological barriers that distinguish the site of administration from the site of action. Ideally, in these situations, the local concentration of the agent at the site(s) of the disease should be high, while the concentration of the agent at the non-target organs and tissues should be lower than certain minimum levels to avoid any adverse reaction.
Drug targeting is known in a very general sense as the capacity of the API to accumulate selectively and quantitatively in the target organ or tissue, regardless of the location and process of administration [19]. The idea of drug targeting, introduced by Paul Ehrlich at the beginning of the twentieth century, considered a hypothetical “magic bullet” as a two-component object; the first should be able to identify and bind onto the target and the second should induce a therapeutic action.
Targeted drug delivery can be divided into two types: passive and active drug targeting. The concept can be briefly explained and is illustrated in Figure 4. A detailed discussion is available in the following subsection, with specific attention paid to antituberculosis drug delivery.
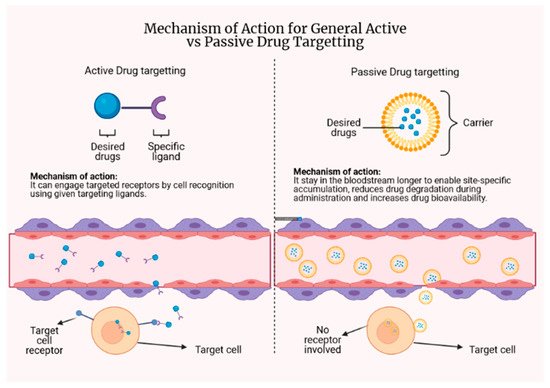
Figure 4. The concept behind active and passive drug targeting.
Various studies are using a passive targeting approach to improve the delivery of antituberculosis agents. Passive targeting relies heavily on the physiological environment of the disease so that there can be a preferential concentration at the site of interest and the limitation of non-specific dissemination. This targeting method involves long-circulating drug delivery systems that remain in the blood to allow site-specific accumulation to occur [19]. The integrated drug should stay in the carrier during circulation in the bloodstream to sufficiently maintain high concentrations at the target sites [20].
The delivery system described under this targeting approach is referred to as a colloidal drug delivery system, as the particles are usually in the colloidal size range (10–400 nm). There are two types of systems developed to date: (1) vesicular delivery systems and (2) particulate delivery systems. The widely researched systems include liposomes (vesicular) and nanoparticles (particulate) [21] with the aim of reducing drug degradation during storage and upon administration, preventing undesirable side effects, and increasing the bioavailability of the drug and the drug fraction accumulated in the pathological region [22]. They have been shown to mediate targeted delivery of pharmaceuticals through various endogenous and/or exogenous physical influences.
The enhanced permeability and retention (EPR) effect makes it easier to distribute particular ligand-modified drugs and drug carriers to poorly accessible areas in a targeted manner [23]. The EPR effect is the mechanism where nanoparticles and macromolecules accumulate more in tumors than in normal tissues due to increased vascular permeability and decreased lymphatic clearance [24]. Since tissues infected by bacteria can also cause increased vascular permeability through inflammation, the EPR effect is stipulated to occur in such tissues [25]. Nanoparticles with longer half-lives would hence have a higher chance to be retained at infected tissues [26].
Nanoparticles have been under the limelight for many years in the search for novel antituberculosis treatments. In this approach, the application of nanoparticles is to target infected macrophages because of nanoparticles are often ingested by circulating macrophages in the blood circulation through the reticuloendothelial system. Because of the natural propensity of the macrophages to engulf particles, passive targeting for extrapulmonary TB has garnered significant attention [27][28]. The differences between normal tissues and pathological cells (infected by Mtb) in terms of pH (acidosis) and temperature (hyperthermia) may induce the release of encapsulated drug in the nanoparticles [29][30]. With this in mind, it was proposed that different pharmaceutical agents be loaded onto pH- or temperature-responsive drug carriers that can alter their properties and release an encapsulated agent when taken to areas with a lower pH or higher temperatures [19]. In addition, the attachment of pH-sensitive polymers onto the nanoparticles surface may also help to induce destabilization and subsequently drug release in specific pH-value compartments.
Targeted delivery of drugs to macrophages has been recognized as a potentially effective strategy [31]. Nanoparticles introduced in the blood circulation will be rapidly taken up by macrophages residing in the host’s filtration organs such as the liver, spleen, and the lung through phagocytosis or endocytosis as part of the host’s inflammatory and immunological responses [32]. Since Mtb resides and multiplies in alveolar macrophages and macrophages are part of the immune cells that will be deployed constantly to infection sites, antituberculosis drugs encapsulated in nanoparticles can be accumulated at the site of infection in a steady manner [33].
Passive targeting of antituberculosis drugs through inhalational routes has a particular significance, as nanoparticles have a sufficient size to enter the alveoli and meet the alveolar macrophages and phagocytoses if their size is less than 5μm. A study reported that the use of a liposome-based formulation of RIF and INH developed for lung delivery was effectively delivered, and both drugs were present in the lungs and alveolar macrophages until day 5 after nebulization. A single liposomal drug nebulization could reach the therapeutic drug levels in 45 min and up to 48 h against unencapsulated drugs that were cleared after 24 h, suggesting a potential reduction in the frequency of daily administration of the two first-line antituberculosis drugs [34]. In addition, the incorporation of negatively charged lipids such as dicetylphosphate (DCP) may also help to increase the ability of alveolar macrophages to capture anionic liposomes via scavenger receptors [35].
In another formulation prepared for intravenous administration of antituberculosis drugs, a multilamellar vesicle made of dipalmitoylphosphatidylcholine (DPPC)/cholesterol (7:2 molar ratio) with PZA was administered to mice with disseminated TB. When administered 2 days a week, efficacy was demonstrated in a total of seven treatment doses, reducing the number of Mtb bacilli in the lungs to a level lower than the control group that received a free drug administered at the same frequency. A more favorable histopathological examination of the lung was also established but less favorable than the free drug administered 6 days/week. Liposome-encapsulated PZA demonstrated advantages over the free drug, but the dose would need proper adjustment and to be regulated accordingly [36]. Micelle-based drug delivery for passive targeting, on the other hand, is typically associated with premature drug release from the micelles and difficulties of delivering intracellular drugs.
Raja et al. compared RIF-loaded controlled-release poly lactic acid-co-glycolic acid (PLGA) nanoparticles against free RIF, and discovered the nanoparticles loaded with RIF are more effective in killing Mycobacterium bovis BCG in macrophages [37]. A similar study by Rani et al. with PEG-PLA (polyethylene glycol-poly-L-lactic acid) polymeric micelle loaded with INH and RIF revealed that the prepared micelles possessed much lower minimum inhibitory concentration (MIC) values for Mtb than free drugs, in addition to being less hemolytic [38]. Niosomes, non-ionic surfactant vesicles loaded with EMB, were prepared by El-Ridy et al. and, following subcutaneous administration, the formulation showed a sustained release pattern of EMB in the lung of Swiss Albino mice [39].
There are also efforts to reformulate second line and prospective antituberculosis drugs. Matteis and colleagues reported the successful encapsulation of bedaquiline, a second line antituberculosis drug with lipid nanoparticles and chitosan-based nano capsules. The nano formulations were found to have a similar MIC as free bedaquiline against Mtb H37Rv and showed no cytotoxic effect on A549, HepG2, and THP-1 cell-lines at MIC [40]. Miranda and her colleagues developed an inhalable multifunctional microparticulate dry powder system using P3, an antituberculosis drug candidate. Upon loading into gelatin and BSA-based microparticles incorporated with superparamagnetic iron oxide nanoparticles, P3 can be released on demand with the use of an external alternate magnetic field, making it possible to release a certain amount of drug at a specific time at the most suitable intervals [41]. In another study by Machelart et al., 2019, ethionamide was successfully co-loaded with a booster molecule BDM43266 in cross-linked poly β-cyclodextrin (pβCD) and administered through an endotracheal route. The reduction of the bacterial load was significant as compared with the untreated group and this showed the ability of pβCD to deliver sufficient concentrations of the drugs in vivo [42].
Encapsulation in nanoparticles has also enabled the simultaneous delivery of multiple drugs that could act synergistically against the bacteria. As an example, Rossi et al. developed spray-dried respirable microparticles from low molecular weight hyaluronic acid sub-micron particles loaded with RIF and INH, as well as verapamil, an efflux pump inhibitor. The microparticles reduced the number of viable bacteria by more than 80% in both susceptible and drug-resistant strains of Mtb. No significant changes occurred when the same microparticles without verapamil were tested against both strains above [43]. The presence of verapamil further helped to improve the efficiency of RIF and INH in bactericidal activity. The activity can potentially reduce the number of pills or injections to be taken, whilst providing a promising treatment outcome.
In active targeting, the delivery of a drug to targeted sites is based on molecular recognition using specific ligands conjugated to the drug, which bind on appropriate receptors expressed by cells at the targeted site. The approach of active targeting enables the administration of drugs at a lower effective dose. There will be fewer drugs being unnecessarily distributed to other sites, hence lowering the quantity of drugs needed to exert a similar effect in comparison with the same drug without a targeting ligand.
Recognition of the target can be carried out at different levels, such as at the level of an entire organ, certain cells specific to that organ, or even individual components of these cells (cell surface antigens). Molecular recognition is undoubtedly the universal type of target recognition since certain components can be found specific to an organ or tissue [19]. The involvement of charged lipids or ligand bindings such as proteins, peptides, polysaccharides, glycolipids, glycoproteins, and monoclonal antibodies is essential to deliver the drug to pathological sites or to cross biological barriers to ensure specific delivery of the drug [27].

