Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anne-Marie Lund Winther | + 2432 word(s) | 2432 | 2021-07-15 04:46:59 | | | |
| 2 | Peter Tang | Meta information modification | 2432 | 2021-08-01 05:46:52 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Winther, A.L. Lipoprotein Lipase Regulation in Atherosclerosis. Encyclopedia. Available online: https://encyclopedia.pub/entry/12630 (accessed on 07 February 2026).
Winther AL. Lipoprotein Lipase Regulation in Atherosclerosis. Encyclopedia. Available at: https://encyclopedia.pub/entry/12630. Accessed February 07, 2026.
Winther, Anne-Marie Lund. "Lipoprotein Lipase Regulation in Atherosclerosis" Encyclopedia, https://encyclopedia.pub/entry/12630 (accessed February 07, 2026).
Winther, A.L. (2021, July 31). Lipoprotein Lipase Regulation in Atherosclerosis. In Encyclopedia. https://encyclopedia.pub/entry/12630
Winther, Anne-Marie Lund. "Lipoprotein Lipase Regulation in Atherosclerosis." Encyclopedia. Web. 31 July, 2021.
Copy Citation
Lipoprotein lipase (LPL) plays a major role in the lipid homeostasis mainly by mediating the intravascular lipolysis of triglyceride rich lipoproteins. Impaired LPL activity leads to the accumulation of chylomicrons and very low-density lipoproteins (VLDL) in plasma, resulting in hypertriglyceridemia. While low-density lipoprotein cholesterol (LDL-C) is recognized as a primary risk factor for atherosclerosis, hypertriglyceridemia has been shown to be an independent risk factor for cardiovascular disease (CVD) and a residual risk factor in atherosclerosis development.
lipoprotein lipase
ANGPTL
atherosclerosis
1. Introduction
There are three main lipids in the blood: cholesterol, phospholipids, and triglycerides. Cholesterol is important for the synthesis of bile acids and steroids and for maintaining the integrity of cell membranes, while phospholipids are a major component of all cell membranes. Triglycerides (TGs) serve as a storable high-energy fuel. In humans, almost 80% of the energy requirements of the heart and liver are fulfilled by oxidation of the long fatty acyl chains present in TGs [1].
Because lipids are poorly soluble in blood plasma, they need to be transported as lipoproteins. Lipoproteins are composed of cholesteryl esters and TGs surrounded by a hydrophilic shell made of phospholipids, unesterified cholesterol, and apolipoproteins. Depending upon the origin of the lipoproteins they differ in size, density, and composition [2][3]. The lipoproteins are generally divided into five major classes. These classes are chylomicrons, VLDL, LDL, intermediate-density lipoprotein (IDL), and high-density lipoproteins (HDL). Each class of lipoprotein serves distinct roles in the lipid metabolism (Figure 1); (i) the large chylomicrons and VLDLs are responsible for the transport and delivery of energy rich TGs, (ii) LDL deposits cholesterol in tissues, and (iii) HDL absorbs cholesterol and transports it back to the liver for degradation and redistribution (Figure 1). The lipoproteins constantly undergo enzymatic processing, exchange of lipids or apolipoproteins, and progressive degradation by lipid unloading. Combined, this network is crucial for lipid homeostasis, and even minor alternations in either of the lipoproteins classes potentially perturbs the entire lipoprotein processing network. LDL and HDL have been found to be important for the development of arteriosclerosis due to their cholesterol cargo. However, studies have shown that elevated plasma triglyceride levels known as hypertriglyceridemia (HTG) is associated with an increased risk of atherosclerosis and CVD [4][5][6]. LPL is responsible for the hydrolysis of the triglyceride in capillaries and thereby transform chylomicrons and VLDL to smaller lipoproteins particles. This process is tightly regulated by a number of proteins that either display a stabilizing, activating, or inhibitory effect of LPL and perturbations of this homeostasis may have anti-atherogenic effects [7][8][9][10][11]. On the other hand, data have shown that when LPL is expressed by macrophages in the vessel wall it displays pro-atherogenic effects [12]. With this in mind, there is a great need to understand the underlining molecular mechanism(s) causing increased triglyceride levels in hypertriglyceridemia and atherosclerosis and map the complex regulation of LPL. This knowledge will likely identify new avenues for lipid lowering strategies that could serve as supplements to the existing interventions (e.g., statins) to help further restraining ASCVD progression.
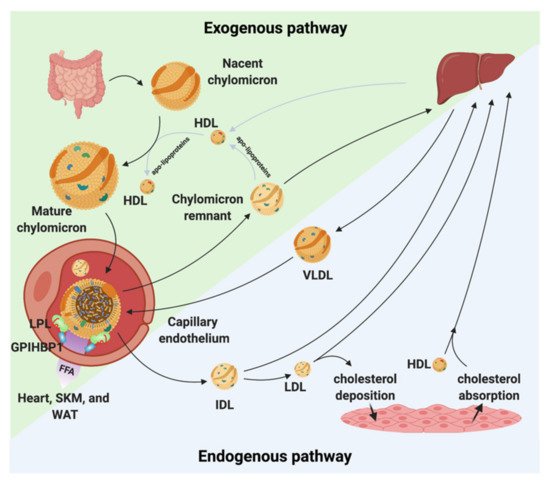
Figure 1. Chylomicron and VLDL life cycle. Chylomicrons are secreted from the intestines to the circulation where they extract apolipoproteins from HDL. The mature chylomicrons are arrested at the capillary endothelium by LPL that is in complex with glycosylphosphatidylinositol anchored high density lipoprotein binding protein 1 (GPIHBP1). LPL performs lipolysis on the encapsulated TGs, which releases free fatty acids that are taken up by the underlying tissues (white adipose tissue (WAT), skeletal muscle (SKM), and the heart). Upon complete hydrolysis, a chylomicron remnant particle is released to the circulation. The chylomicron remnant particle back-exchanges apolipoproteins to circulating HDL, before final catabolism in the liver. A similar life cycle is found for VLDL. VLDL is secreted from the liver and is arrested and hydrolyzed by the LPL•GPIHBP1 complex. This releases IDL, which can either be taken up by the liver or be hydrolyzed further into LDL. LDL deposits cholesterol esters in various tissues. HDL absorbs cholesterol and transports it to the liver.
2. The Lipoprotein Lipase
The central role of LPL in triglyceride hydrolysis was recognized over 60 years ago by the discovery of the enzyme in 1955 [13]. The expression level of LPL is robust in cells and organs with a high oxidative metabolism, but it is also expressed in other tissue types, not related to intravascular lipolysis such as the spleen, testis, lung, kidneys, and brain as well as in macrophages [14]. LPL is a 55 kDa glycoprotein consisting of an N-terminal α/β-hydrolase domain and a C-terminal Polycystin-1, Lipoxygenase, Alpha-Toxin (PLAT) domain (Figure 2). The α/β-hydrolase domain harbors the active site that primarily hydrolyses the sn-1/sn-3 ester bonds of triglycerides, thereby releasing two unesterified fatty acids and a sn-2 monoacylglycerol [15]. The hydrolysis takes place at the luminal face of the capillary endothelium where LPL hydrolyzes the triglycerides within the neutral cores of chylomicrons and VLDLs producing chylomicron remnants and IDLs, respectively (Figure 1) [16]. The released free fatty acids (FFA) can either be used as an energy source or be re-esterified and stored in adipose tissues as triglycerides.
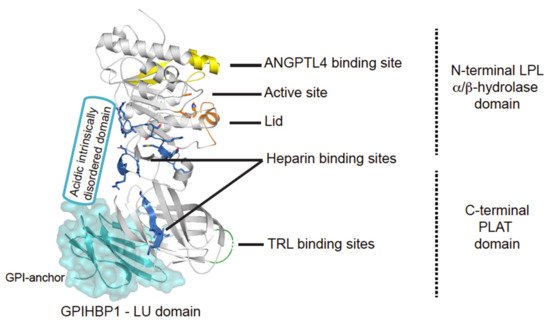
Figure 2. Structural elements in LPL. Cartoon representation of the human LPL•GPIHBP1 complex and with the molecular surface of the LU domain (Ly6/uPAR protein domain family) of GPIHBP1 shown as a transparent light blue envelope (generated with The PyMol Molecular Graphic System (Version 2.0 Schrödinger, LLC) using coordinates from the LPL•GPIHBP1 crystal structure; Protein Data Bank ID code 6E7K). LPL elements implicated in ANGPTL4 binding are highlighted in yellow [17]. GPIHBP1 binds to the C-terminal PLAT domain of LPL, and the location of GPIHBP1’s membrane-tethering site (GPI anchor) is indicated [18]. GPIHBP1’s acidic intrinsically disordered domain stabilizes LPL’s α/β-hydrolase domain and is assumed to interact with a large basic patch on the surface of LPL [19][20]. The active site containing the catalytic triad (Ser134, Asp158, and His243) is represented with orange residues [21] and the orange helix is the lid covering the active site responsible for substrate specificity [22]. The triglyceride-rich lipoprotein (TRL) binding site is indicated in green and residues important for heparin binding is shown in blue [23][24][25].
3. LPL’s Activity Is Regulated by Apolipoproteins
Apolipoproteins are integrated in the maintenance of lipid homeostasis. They constitute a class of amphipathic proteins having a hydrophobic region that interacts with lipids in the lipoprotein particles, while the hydrophilic region allows interaction with soluble proteins. They provide structure-defining elements for lipoprotein particles and serve as ligands for specific receptors. Both ApoC-II and ApoA-V act as positive regulators of LPL in intravascular lipolysis, while ApoC-I and ApoC-III display an inhibitory role towards LPL. Apolipoproteins may promote atherogenic or anti-atherogenic effects depending on their expression levels and microenvironment.
4. Regulation of LPL Lipolytic Activity by ANGPTLs
Intravascular lipolysis is strictly regulated by specific moderators of LPL activity. A tight temporal and spatial regulation of LPL ensures that the flux of FFA arrives at the right time in target tissues in need of energy supply. This anatomical regulation is mediated by members of the angiopoietin-like (ANGPTL) family, ANGPTL3, ANGPTL4, and ANGPTL8 (Figure 3) [26]. ANGPTL3 and ANGPTL4 are both composed by an N-terminal coiled-coil domain (CCD), a linker region, and a C-terminal fibrinogen-like domain (FLD), while ANGPTL8 lacks a FLD. ANGPTL3 and ANGPLT4 both inhibit LPL, but with vastly different efficacy, and recent studies have expanded the complexity of LPL regulation as ANGPTL8 binding increased the potency of ANGPTL3 and reduces that of ANGPLT4. Like other key players in TG metabolism, the atherosclerotic profiles for ANGPTLs are context dependent, highlighting the importance of understanding their biological and biochemistry.
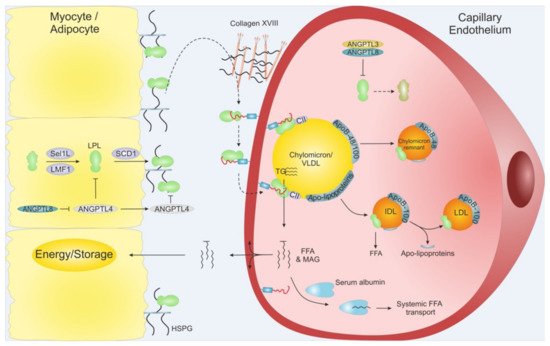
Figure 3. Schematic overview of LPL translocation. LPL is secreted from adipose tissue and myocytes from skeletal muscles and heart. Intracellular LPL folding and secretion is assisted by LMF1, Sel1L and SCD1. After secretion, active LPL is bound to cell surface HSPG before being translocated to GPIHBP1, potentially via Collagen XVIII. GPIHBP1 captures LPL in the subendothelial space and mediates LPL trancytosis across the capillary endothelium, into the lumen. Within the lumen, the LPL•GPIHBP1 complex is responsible for the arrest of VLDLs and chylomicrons. After final hydrolysis, either a chylomicron remnant particle or an IDL are released to be taken up by the liver or turned into LDL via further TG hydrolysis, respectively. ANGPTL4 act as a potent LPL inhibitor in the adipose tissue, and ANGPTL3•ANGPTL8 act on LPL mainly in the capillaries of the muscles and heart. The inhibition of the ANGPTLs is driven by nutritional status and exercise. MAG: Monoacylglycerol.
5. Dysfunctional LPL and Hypertriglyceridemia
Homozygous or compound heterozygous individuals with loss-of-function mutations in LPL, ApoC-II, ApoA-V, LMF1, and GPIHBP1 develop a rare monogenic disorder known as familial chylomicronaemia syndrome (FCS). The disorder is defined by an excessive accumulation of chylomicrons with clinical symptoms such as eruptive xanthomas and acute pancreatitis that are associated with very high serum TGs concentrations (>2000 mg/dL) [27][28][29]. The more common polygenic variant of HTG is caused by genetic factors in combination with secondary factors such as type 2 diabetes, obesity, renal disease, pregnancy, and alcohol intake and is characterized by increased plasma levels of TRLs and reduced levels of HDL-C [30]. GWAS identified an association between 32 common gene variants and elevated plasma TGs concentrations and in many cases increased risk of CVD. These variants include ApoA-V, LPL, and ApoB, which are all strong determinants of polygenic HTG [31]. Of the more than 100 LPL variants that have been identified, a subset has been classified as pathogenic. The more common LPL variants (LPL-N291A, LPL-D9N, and LPL-G188E) are associated with mildly increased TGs levels [32] and generate an atherogenic lipoprotein profile [5]. On the contrary, another common LPL variant with gain-of-function characteristics (LPL-S447X), is associated with decreased plasma TGs levels compared to non-carriers [32] and lower risk of CHD [33]. The beneficial properties of this variant were exploited in a LPL gene therapy strategy (Alipogene tiparvovec) for treatment of patients with LPL deficiency. The trials demonstrated transiently reduced fasting plasma triglyceride levels compared to baseline after 12 weeks of administration, and reduction in pancreatitis incidences [34]. Despite these promising results, Alipogene tiparvovec was discontinued as therapy due to excessive costs. Currently, first line of treatment for HTG is dietary and lifestyle interventions and control of secondary factors. Recent developments have nonetheless provided new treatment options for patients with monogenic HTG as exemplified by the approval of Volanesorsen, an ApoC-III targeting drug, for the treatment of adults with FCS [35].
6. Clearance of Processed Lipoproteins and Risk of Atherosclerosis
After intravascular processing of the marginated TRLs, the remaining lipoprotein (remnant particle) detaches from the endothelial cells along with some inactive LPL molecules still attached. That inactive LPL acts as a ligand for a number of internalization receptors present on hepatocytes, facilitating the hepatic uptake of remnant particles [36][37]. These receptors include LDL receptor, VLDL receptor, LRP1 and SCD1 [38][39][40][41][42][43][44]. A possible downside of this mechanism, is that, LPL may act to promote LDL and VLDL retention in the subendothelial cell matrix thus delaying their return to the circulation by endothelial transcytosis [45]. Such LPL-mediated LDL retention represents a possible pathogenic factor for the progression of the atherosclerotic lesions as it promotes lipoprotein uptake in macrophages increasing the risk of foam cell formation and inflammation [12]. In the arterial intima, LPL facilitates this temporal sequestering of lipoproteins by its strong ionic interactions with proteoglycans such as biglycan, versican, and collagen XVIII [46][47]. Excessive accumulation of lipoproteins in the subendothelial space leads to fatty streaks, which is a hallmark of early precursor lesions in atherogenesis leading to plaque formation and progression [48][49]. When macrophages are recruited to lipid-laden precursor lesions in the arterial intima, they start phagocytosing the dense lipid depositions and transform into foam cells. This elicits a number of pathogenic responses ultimately leading to chronic inflammation: (i) excessive lipid overload in macrophages impairs cholesterol efflux [50] and (ii) secretion of pro-inflammatory cytokines and ROS upon macrophage activation by oxLDL [51][52]. Furthermore, these foam cells secrete a number of proteases that degrade the extracellular matrix weakening the plaque structure provoking its rupture with severe cardiovascular damage and risk of thromboembolism [53][54]. Studies in mice show that macrophages display increased LPL expression in early atherosclerosis [55] and the enhanced LPL expression accelerates foam cell formation [56][57]. Importantly, mice with Lpl−/− confined to macrophages have reduced lesion sizes and lower diet-induced atherosclerosis [58][59].
7. Remnant Particles and Their Role in Atherosclerosis
Very severe HTG characterized by the accumulation of chylomicrons and large VLDLs is generally not considered pro-atherogenic since these particles are too large to be trancytosed across endothelial cells and enter the arterial intima. Remnant lipoproteins, however, with a diameter <70 nm can be transcytosed [60]. IDL and small VLDL particles readily enter the arterial intima and are hence considered atherogenic [61]. The high atherogenic impact of VLDLs and IDLs in arterial intima is underscored by the finding that the majority of ApoB isolated from human atherosclerotic plaques is derived from VLDL and IDL lipoproteins rather than LDL [62]. Studies on hyperlipidemic rabbits show that both VLDL and IDL particles enter and are retained in the arterial intima to the same extent as LDL particles [63]. Other studies claim remnant particles to be more atherogenic than LDL itself [64]. This proposition is underscored by two observations. First, remnant particles do not need to be oxidized before they are taken up by macrophages in the arterial intima [64][65]. Second, they carry approximately 40 times more cholesterol per remnant particle compared to LDL particles [60]. Previously, oxLDL and chylomicron remnants were considered the main atherogenic lipoproteins, but VLDL remnants may have an even greater atherogenic potential [66].
The formation of remnant particles is an integral part of intravascular lipolysis and represents a byproduct of normal lipid homeostasis. Therapeutic interventions preventing the retention of remnant particles in the arterial intima appear more straightforward than limiting the production of remnant particles. One such therapeutic approach is to block or reduce binding of ApoB to HSPGs in the subendothelial spaces. ApoB exists in two isoforms on lipoprotein particles, ApoB-48 and ApoB-100, both of which are encoded by the same gene and play a crucial role in lipid homeostasis [67]. ApoB-100 is a very large protein containing 4536 amino acids [68]. It is expressed in liver and all lipoproteins synthesized by hepatocytes (VLDL) contain ApoB-100 [68]. On the other hand, the shorter isoform ApoB-48 is found on lipoproteins of intestinal origin [69]. ApoB-100 is required for the synthesis, assembly and secretion of TRLs originating from liver. Loss-of-function variants in APOB cause hypobetalipoproteinemia, normotriglyceridemic hypobetalipoproteinemia, and hypercholesterolemia—diseases affecting plasma cholesterol and ApoB levels. In aggregate, these observations highlight the potential of ApoB as a clinical biomarker in diagnostics as well as therapeutic target. Mipomersen is an FDA-approved second generation antisense oligonucleotide, that has been designed to reduce plasma concentration of ApoB-100 [70]. Since ApoB-100 is an integrated component of all atherogenic lipoproteins (including LDL), use of Mipomersen may also reduce the risk of atherosclerosis.
References
- Cox, M.; Nelson, D.L. Lehninger Principles of Biochemistry, 4th ed; W.H. Freeman: New York, NY, USA, 2005; ISBN 0716743396.
- Bayly, G.R. Lipids and disorders of lipoprotein metabolism. In Clinical Biochemistry: Metabolic and Clinical Aspects; Elsevier: New York, NY, USA, 2014; pp. 702–736. ISBN 9780702054785.
- Nordestgaard, B.G. A Test in Context: Lipid Profile, Fasting Versus Nonfasting. J. Am. Coll. Cardiol. 2017, 70, 1637–1646.
- Hokanson, J.E.; Austin, M.A. Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: A metaanalysis of population-based prospective studies. Eur. J. Cardiovasc. Prev. Rehabil. 1996, 3, 213–219.
- Wittrup, H.H.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Lipoprotein lipase mutations, plasma lipids and lipoproteins, and risk of ischemic heart disease: A meta-analysis. Circulation 1999, 99, 2901–2907.
- Sarwar, N.; Danesh, J.; Eiriksdottir, G.; Sigurdsson, G.; Wareham, N.; Bingham, S.; Boekholdt, S.M.; Khaw, K.T.; Gudnason, V. Triglycerides and the risk of coronary heart disease: 10 158 Incident cases among 262 525 participants in 29 Western prospective studies. Circulation 2007, 115, 450–458.
- Ando, Y.; Shimizugawa, T.; Takeshita, S.; Ono, M.; Shimamura, M.; Koishi, R.; Furukawa, H. A decreased expression of angiopoietin-like 3 is protective against atherosclerosis in apoE-deficient mice. J. Lipid Res. 2003, 44, 1216–1223.
- Stitziel, N.O.; Khera, A.V.; Wang, X.; Bierhals, A.J.; Vourakis, A.C.; Sperry, A.E.; Natarajan, P.; Klarin, D.; Emdin, C.A.; Zekavat, S.M.; et al. ANGPTL3 Deficiency and Protection Against Coronary Artery Disease. J. Am. Coll. Cardiol. 2017, 69, 2054–2063.
- Dewey, F.E.; Gusarova, V.; O’Dushlaine, C.; Gottesman, O.; Trejos, J.; Hunt, C.; Van Hout, C.V.; Habegger, L.; Buckler, D.; Lai, K.-M.V.; et al. Inactivating Variants in ANGPTL4 and Risk of Coronary Artery Disease. N. Engl. J. Med. 2016, 374, 1123–1133.
- Smart-Halajko, M.C.; Kelley-Hedgepeth, A.; Montefusco, M.C.; Cooper, J.A.; Kopin, A.; McCaffrey, J.M.; Balasubramanyam, A.; Pownall, H.J.; Nathan, D.M.; Peter, I.; et al. ANGPTL4 variants E40K and T266M are associated with lower fasting triglyceride levels in Non-Hispanic White Americans from the Look AHEAD Clinical Trial. BMC Med. Genet. 2011, 12, 89.
- Dewey, F.E.; Gusarova, V.; Dunbar, R.L.; O’Dushlaine, C.; Schurmann, C.; Gottesman, O.; McCarthy, S.; Van Hout, C.V.; Bruse, S.; Dansky, H.M.; et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 211–221.
- Gustafsson, M.; Levin, M.; Skålén, K.; Perman, J.; Fridén, V.; Jirholt, P.; Olofsson, S.O.; Fazio, S.; Linton, M.F.; Semenkovich, C.F.; et al. Retention of low-density lipoprotein in atherosclerotic lesions of the mouse: Evidence for a role of lipoprotein lipase. Circ. Res. 2007, 101, 777–783.
- Korn, E.D. Clearing factor, a Heparin-activated lipoprotein lipase: II. Substrate Specificity and activation of coconut oil. J. Biol. Chem. 1955, 215, 15–26.
- Kirchgessner, T.G.; LeBouef, R.C.; Langner, C.A.; Zollman, S.; Chang, C.H.; Taylor, B.A.; Schotz, M.C.; Gordon, J.I.; Lusis, A.J. Genetic and developmental regulation of the lipoprotein lipase gene: Loci both distal and proximal to the lipoprotein lipase structural gene control enzyme expression. J. Biol. Chem. 1989, 264, 1473–1482.
- Scow, R.O.; Olivecrona, T. Effect of albumin on products formed from chylomicron triacylglycerol by lipoprotein lipase in vitro. Biochim. Biophys. Acta Lipids Lipid Metab. 1977, 487, 472–486.
- Brunzell, J.D.; Hazzard, W.R.; Porte, D.; Bierman, E.L. Evidence for a common, saturable, triglyceride removal mechanism for chylomicrons and very low density lipoproteins in man. J. Clin. Investig. 1973, 52, 1578–1585.
- Leth-Espensen, K.Z.; Kristensen, K.K.; Kumari, A.; Winther, A.-M.L.; Young, S.G.; Jørgensen, T.J.D.; Ploug, M. The intrinsic instability of the hydrolase domain of lipoprotein lipase facilitates its inactivation by ANGPTL4-catalyzed unfolding. Proc. Natl. Acad. Sci. USA 2021, 118, e2026650118.
- Birrane, G.; Beigneux, A.P.; Dwyer, B.; Strack-Logue, B.; Kristensen, K.K.; Francone, O.L.; Fong, L.G.; Mertens, H.D.T.; Pan, C.Q.; Ploug, M.; et al. Structure of the lipoprotein lipase–GPIHBP1 complex that mediates plasma triglyceride hydrolysis. Proc. Natl. Acad. Sci. USA 2019, 116, 1723–1732.
- Kristensen, K.K.; Midtgaard, S.R.; Mysling, S.; Kovrov, O.; Hansen, L.B.; Skar-Gislinge, N.; Beigneux, A.P.; Kragelund, B.B.; Olivecrona, G.; Young, S.G.; et al. A disordered acidic domain in GPIHBP1 harboring a sulfated tyrosine regulates lipoprotein lipase. Proc. Natl. Acad. Sci. USA 2018, 115, E6020–E6029.
- Mysling, S.; Kristensen, K.K.; Larsson, M.; Beigneux, A.P.; Gårdsvoll, H.; Fong, L.G.; Bensadouen, A.; Jørgensen, T.J.; Young, S.G.; Ploug, M. The acidic domain of the endothelial membrane protein GPIHBP1 stabilizes lipoprotein lipase activity by preventing unfolding of its catalytic domain. Elife 2016, 5, e12095.
- Emmerich, J.; Beg, O.U.; Peterson, J.; Previato, L.; Brunzell, J.D.; Brewer, H.B.; Santamarina-Fojo, S. Human lipoprotein lipase. Analysis of the catalytic triad by site-directed mutagenesis of Ser-132, Asp-156, and His-241. J. Biol. Chem. 1992, 267, 4161–4165.
- Dugi, K.A.; Dichek, H.L.; Talley, G.D.; Brewer, H.B.; Santamarina-Fojo, S. Human lipoprotein lipase: The loop covering the catalytic site is essential for interaction with lipid substrates. J. Biol. Chem. 1992, 267, 25086–25091.
- Ma, Y.; Henderson, H.E.; Liu, M.S.; Zhang, H.; Forsythe, I.J.; Clarke-Lewis, I.; Hayden, M.R.; Brunzell, J.D. Mutagenesis in four candidate heparin binding regions (residues 279–282, 291–304, 390–393, and 439–448) and identification of residues affecting heparin binding of human lipoprotein lipase. J. Lipid Res. 1994, 35, 2049–2059.
- Lookene, A.; Nielsen, M.S.; Gliemann, J.; Olivecrona, G. Contribution of the carboxy-terminal domain of lipoprotein lipase to interaction with heparin and lipoproteins. Biochem. Biophys. Res. Commun. 2000, 271, 15–21.
- Lutz, E.P.; Merkel, M.; Kako, Y.; Melford, K.; Radner, H.; Breslow, J.L.; Bensadoun, A.; Goldberg, I.J. Heparin-binding defective lipoprotein lipase is unstable and causes abnormalities in lipid delivery to tissues. J. Clin. Investig. 2001, 107, 1183–1192.
- Santulli, G. Angiopoietin-like proteins: A comprehensive look. Front. Endocrinol. 2014, 5, 5–10.
- Brahm, A.; Hegele, R.A. Hypertriglyceridemia. Nutrients 2013, 5, 981.
- Simha, V. Management of hypertriglyceridemia. BMJ 2020, 371, m3109.
- Surendran, R.P.; Visser, M.E.; Heemelaar, S.; Wang, J.; Peter, J.; Defesche, J.C.; Kuivenhoven, J.A.; Hosseini, M.; Péterfy, M.; Kastelein, J.J.P.; et al. Mutations in LPL, APOC2, APOA5, GPIHBP1 and LMF1 in patients with severe hypertriglyceridaemia. J. Intern. Med. 2012, 272, 185–196.
- Yuan, G.; Al-Shali, K.Z.; Hegele, R.A. Hypertriglyceridemia: Its etiology, effects and treatment. CMAJ 2007, 176, 1113–1120.
- Johansen, C.T.; Hegele, R.A. Allelic and phenotypic spectrum of plasma triglycerides. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2012, 1821, 833–842.
- Rodrigues, R.; Artieda, M.; Tejedor, D.; Martínez, A.; Konstantinova, P.; Petry, H.; Meyer, C.; Corzo, D.; Sundgreen, C.; Klor, H.U.; et al. Pathogenic classification of LPL gene variants reported to be associated with LPL deficiency. J. Clin. Lipidol. 2016, 10, 394–409.
- Gagné, S.E.; Larson, M.G.; Pimstone, S.N.; Schaefer, E.J.; Kastelein, J.J.P.; Wilson, P.W.F.; Ordovas, J.M.; Hayden, M.R. A common truncation variant of lipoprotein lipase (Ser447X) confers protection against coronary heart disease: The Framingham Offspring Study. Clin. Genet. 1999, 55, 450–454.
- Gaudet, D.; Méthot, J.; Kastelein, J. Gene therapy for lipoprotein lipase deficiency. Curr. Opin. Lipidol. 2012, 23, 310–320.
- Paik, J.; Duggan, S. Volanesorsen: First Global Approval. Drugs 2019, 79, 1349–1354.
- Felts, J.M.; Itakura, H.; Thomas Crane, R. The mechanism of assimilation of constituents of chylomicrons, very low density lipoproteins and remnants—A new theory. Biochem. Biophys. Res. Commun. 1975, 66, 1467–1475.
- Merkel, M.; Heeren, J.; Dudeck, W.; Rinninger, F.; Radner, H.; Breslow, J.L.; Goldberg, I.J.; Zechner, R.; Greten, H. Inactive lipoprotein lipase (LPL) alone increases selective cholesterol ester uptake in vivo, whereas in the presence of active LPL it also increases triglyceride hydrolysis and whole particle lipoprotein uptake. J. Biol. Chem. 2002, 277, 7405–7411.
- Beisiegel, U.; Weber, W.; Bengtsson-Olivecrona, G. Lipoprotein lipase enhances the binding of chylomicrons to low density lipoprotein receptor-related protein. Proc. Natl. Acad. Sci. USA 1991, 88, 8342–8346.
- Takahashi, S.; Kawarabayasi, Y.; Nakai, T.; Sakai, J.; Yamamoto, T. Rabbit very low density lipoprotein receptor: A low density lipoprotein receptor-like protein with distinct ligand specificity. Proc. Natl. Acad. Sci. USA 1992, 89, 9252–9256.
- Mulder, M.; Lombardi, P.; Jansen, H.; van Berkel, T.J.C.; Frants, R.R.; Havekes, L.M. Heparan sulphate proteoglycans are involved in the lipoprotein lipase-mediated enhancement of the cellular binding of very low density and low density lipoproteins. Biochem. Biophys. Res. Commun. 1992, 185, 582–587.
- Mulder, M.; Lombardi, P.; Jansen, H.; Van Berkel, T.J.C.; Frants, R.R.; Havekes, L.M. Low density lipoprotein receptor internalizes low density and very low density lipoproteins that are bound to heparan sulfate proteoglycans via lipoprotein lipase. J. Biol. Chem. 1993, 268, 9369–9375.
- Chappell, D.A.; Fry, G.L.; Waknitz, M.A.; Muhonen, L.E.; Pladet, M.W.; Iverius, P.H.; Strickland, D.K. Lipoprotein lipase induces catabolism of normal triglyceride-rich lipoproteins via the low density lipoprotein receptor-related protein/α2- macroglobulin receptor in vitro. A process facilitated by cell-surface proteoglycans. J. Biol. Chem. 1993, 268, 14168–14175.
- Nykjaer, A.; Bengtsson-Olivecrona, G.; Lookene, A.; Moestrup, S.K.; Petersen, C.M.; Weber, W.; Beisiegel, U.; Gliemann, J. The α2-macroglobulin receptor/low density lipoprotein receptor-related protein binds lipoprotein lipase and β-migrating very low density lipoprotein associated with the lipase. J. Biol. Chem. 1993, 268, 15048–15055.
- Niemeier, A.; Gåfvels, M.; Heeren, J.; Meyer, N.; Angelin, B.; Beisiegel, U. VLDL receptor mediates the uptake of human chylomicron remnants in vitro. J. Lipid Res. 1996, 37, 1733–1742.
- Saxena, U.; Klein, M.G.; Vanni, T.M.; Goldberg, I.J. Lipoprotein lipase increases low density lipoprotein retention by subendothelial cell matrix. J. Clin. Investig. 1992, 89, 373–380.
- O’Brien, K.D.; Olin, K.L.; Alpers, C.E.; Chiu, W.; Ferguson, M.; Hudkins, K.; Wight, T.N.; Chait, A. Comparison of apolipoprotein and proteoglycan deposits in human coronary atherosclerotic plaques: Colocalization of biglycan with apolipoproteins. Circulation 1998, 98, 519–527.
- Olin, K.L.; Potter-Perigo, S.; Barrett, P.H.R.; Wight, T.N.; Chait, A. Lipoprotein lipase enhances the binding of native and oxidized low density lipoproteins to versican and biglycan synthesized by cultured arterial smooth muscle cells. J. Biol. Chem. 1999, 274, 34629–34636.
- Skålén, K.; Gustafsson, M.; Knutsen Rydberg, E.; Hultén, L.M.; Wiklund, O.; Innerarity, T.L.; Boren, J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 2002, 417, 750–754.
- Joris, I.; Zand, T.; Nunnari, J.J.; Krolikowski, F.J.; Majno, G. Studies on the pathogenesis of atherosclerosis. I. Adhesion and emigration of mononuclear cells in the aorta of hypercholesterolemic rats. Am. J. Pathol. 1983, 113, 341–358.
- Huff, M.W.; Daugherty, A.; Lu, H. Atherosclerosis, 6th ed.; Elsevier: New York, NY, USA, 2016; ISBN 9780444634382.
- Chen, C.; Khismatullin, D.B. Oxidized low-density lipoprotein contributes to atherogenesis via co-activation of macrophages and mast cells. PLoS ONE 2015, 10, e123088.
- Stocker, R.; Keaney, J.F. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478.
- Thomas, A.C.; Sala-Newby, G.B.; Ismail, Y.; Johnson, J.L.; Pasterkamp, G.; Newby, A.C. Genomics of foam cells and nonfoamy macrophages from rabbits identifies arginase-I as a differential regulator of nitric oxide production. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 571–577.
- Nagai, M.; Granger, D.N. Inflammatory Mechanisms in Ischemic Cerebrovascular Disease; Elsevier Inc.: New York, NY, USA, 2018; ISBN 9780128117095.
- Renier, G.; Skamene, E.; De Sanctis, J.B.; Radzioch, D. High macrophage lipoprotein lipase expression and secretion are associated in inbred murine strains with susceptibility to atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 1993, 13, 190–196.
- Babaev, V.R.; Fazio, S.; Gleaves, L.A.; Carter, K.J.; Semenkovich, C.F.; Linton, M.F. Macrophage lipoprotein lipase promotes foam cell formation and atherosclerosis in vivo. J. Clin. Investig. 1999, 103, 1697–1705.
- Babaev, V.R.; Patel, M.B.; Semenkovich, C.F.; Fazio, S.; Linton, M.F. Macrophage lipoprotein lipase promotes foam cell formation and atherosclerosis in low density lipoprotein receptor-deficient mice. J. Biol. Chem. 2000, 275, 26293–26299.
- Takahashi, M.; Yagyu, H.; Tazoe, F.; Nagashima, S.; Ohshiro, T.; Okada, K.; Osuga, J.I.; Goldberg, I.J.; Ishibashi, S. Macrophage lipoprotein lipase modulates the development of atherosclerosis but not adiposity. J. Lipid Res. 2013, 54, 1124–1134.
- Van Eck, M.; Zimmermann, R.; Groot, P.H.; Zechner, R.; Van Berkel, T.J. Role of macrophage-derived lipoprotein lipase in lipoprotein metabolism and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2000, 20.
- Borén, J.; Williams, K.J. The central role of arterial retention of cholesterolrich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: A triumph of simplicity. Curr. Opin. Lipidol. 2016, 27, 473–483.
- Jørgensen, A.B.; Frikke-Schmidt, R.; Nordestgaard, B.G.; Tybjærg-Hansen, A. Loss-of-Function Mutations in APOC3 and Risk of Ischemic Vascular Disease. N. Engl. J. Med. 2014, 371, 32–41.
- Rapp, J.H.; Lespine, A.; Hamilton, R.L.; Colyvas, N.; Chaumeton, A.H.; Tweedie-Hardman, J.; Kotite, L.; Kunitake, S.T.; Havel, R.J.; Kane, J.P. Triglyceride-rich lipoproteins isolated by selected-affinity anti- apolipoprotein B immunosorption from human atherosclerotic plaque. Arterioscler. Thromb. 1994, 14, 1767–1774.
- Nordestgaard, B.G.; Wootton, R.; Lewis, B. Selective retention of VLDL, IDL, and LDL in the arterial intima of genetically hyperlipidemic rabbits in vivo: Molecular size as a determinant of fractional loss from the intima-inner media. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 534–542.
- Nakajima, K.; Nakano, T.; Tanaka, A. The oxidative modification hypothesis of atherosclerosis: The comparison of atherogenic effects on oxidized LDL and remnant lipoproteins in plasma. Clin. Chim. Acta 2006, 367, 36–47.
- Tomono, S.; Kawazu, S.; Kato, N.; Ono, T.; Ishii, C.; Ito, Y.; Shimizu, M.; Shimoyama, M.; Nakano, T.; Nakajima, K. Uptake of remnant like particles (RLP) in diabetic patients from mouse peritoneal macrophages. J. Atheroscler. Thromb. 1994, 1, 98–102.
- Nakajima, K.; Tanaka, A. Atherogenic postprandial remnant lipoproteins; VLDL remnants as a causal factor in atherosclerosis. Clin. Chim. Acta 2018, 478, 200–215.
- Chen, S.H.; Yang, C.Y.; Chen, P.F.; Setzer, D.; Tanimura, M.; Li, W.H.; Gotto, A.M.; Chan, L. The complete cDNA and amino acid sequence of human apolipoprotein B-100. J. Biol. Chem. 1986, 261, 12918–12921.
- Glickman, R.M.; Rogers, M.; Glickman, J.N. Apolipoprotein B synthesis by human liver and intestine in vitro. Proc. Natl. Acad. Sci. USA 1986, 83, 5296–5300.
- Chen, S.; Habib, G.; Yang, C.; Gu, Z.; Lee, B.; Weng, S.; Silberman, S.; Cai, S.; Deslypere, J.; Rosseneu, M.; et al. Apolipoprotein B-48 is the product of a messenger RNA with an organ-specific in-frame stop codon. Science 1987, 238, 363–366.
- Robinson, J.G. Management of Familial Hypercholesterolemia: A Review of the Recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J. Manag. Care Pharm. 2013, 19, 139–149.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.9K
Revisions:
2 times
(View History)
Update Date:
01 Aug 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No