Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Elisa Vicenzi | + 3587 word(s) | 3587 | 2021-07-26 04:43:29 | | | |
| 2 | Bruce Ren | -21 word(s) | 3566 | 2021-07-29 10:52:51 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Vicenzi, E. TRIM22. A Multitasking Antiviral Factor. Encyclopedia. Available online: https://encyclopedia.pub/entry/12571 (accessed on 07 February 2026).
Vicenzi E. TRIM22. A Multitasking Antiviral Factor. Encyclopedia. Available at: https://encyclopedia.pub/entry/12571. Accessed February 07, 2026.
Vicenzi, Elisa. "TRIM22. A Multitasking Antiviral Factor" Encyclopedia, https://encyclopedia.pub/entry/12571 (accessed February 07, 2026).
Vicenzi, E. (2021, July 29). TRIM22. A Multitasking Antiviral Factor. In Encyclopedia. https://encyclopedia.pub/entry/12571
Vicenzi, Elisa. "TRIM22. A Multitasking Antiviral Factor." Encyclopedia. Web. 29 July, 2021.
Copy Citation
Viral invasion of target cells triggers an immediate intracellular host defense system aimed at preventing further propagation of the virus. Viral genomes or early products of viral replication are sensed by a number of pattern recognition receptors, leading to the synthesis and production of type I interferons (IFNs) that, in turn, activate a cascade of IFN-stimulated genes (ISGs) with antiviral functions. Among these, several members of the tripartite motif (TRIM) family are antiviral executors.
TRIM22
DNA and RNA viruses
HIV-1
influenza A virus
interferons
1. Introduction
Innate immunity represents the frontline defense against viruses, aiming at preserving the host from viral invasion. Part of this complex network of cells and soluble factors is the intrinsic capacity of every cell to trigger a set of intracellular responses to viral infection in order to curtail its replicative capacity and further viral spreading. In fact, the cell response to viral entry is rapid and unspecific as both viral RNA and DNA genomes are sensed rapidly after their release in the cytoplasm by exposing evolutionarily conserved pathogen-associated molecular patterns (PAMP) to cellular germline-encoded pattern recognition receptors (PRRs) [1]. Viral component recognition initiates a signaling cascade that ultimately leads to transcription of pro-inflammatory cytokines and of type I interferons (IFNs), namely, IFN-β, firstly, and then IFN-α, the latter actually being a mixture of several proteins [2]. Type I IFNs bind to the IFN-α receptor (IFNAR) [3] in order to induce the expression of hundreds of IFN-stimulated genes (ISGs) that interfere with distinct stages of virus replication [4][5]. Among these ISGs, many TRIM proteins have been described to exert antiviral functions [5][6][7].
More than 80 TRIM proteins have been identified to share common structural features. TRIM proteins are characterized by an RBCC motif composed of an N-terminus domain, followed by a central region with one or two B boxes and a coiled-coil (CC) region. The RBCC motif is flanked by a C-terminus domain [8]. The N-terminus domain, defined as a RING (Really Interesting New Gene), is endowed with E3 ubiquitin ligase activity [9]. The CC domain is characterized by structural features that favor protein–protein interactions with different TRIM family members [10][11], but also other proteins [12]. The C-terminus domain is the most variable region among the TRIM proteins, and it is used to classify them into families [13][14].
TRIM protein members are classified into 11 families (from C-I to C-XI) based on their overall domain structure, with one group of TRIM proteins remaining unclassified due to a lack of a RING domain (e.g., TRIM14 and TRIM20) [13][15]. Many TRIM proteins have an antiviral function, and most of them belong to the C-IV family that represents the largest family with 34 members. This family is characterized by having a SPRY domain, or a SPRY region, in combination with a PRY domain to form a B30.2 domain at the C-terminus following the CC region. The B30.2 domain was originally identified as a protein domain encoded by a single exon (called B30-2) in the human major histocompatibility complex class I (MHC-I) region [16], and in genes involved in autoimmune and genetic diseases [17]. The SPRY domain was identified as a conserved domain in the non-receptor tyrosine kinase spore lysis A (splA) of Dictyostelium discoideum, and in mammalian ryanodine receptors (RyR) [18]. TRIM22 is characterized by a B30.2 domain including PRY and SPRY regions [19].
The member of the TRIM family that has historically received more attention as antiviral factor, particularly as an anti-HIV-1 determinant, is TRIM5α [20]. Interestingly, the TRIM5 gene is located on chromosome 11 adjacent to the TRIM22 gene [21]. Their proximity has been linked to a dynamic history of gene expansion and loss in mammals. For example, the cow genome encodes TRIM5 but has lost TRIM22, and vice versa, the dog genome encodes TRIM22 but has lost TRIM5. In primates, TRIM22 is present, although signatures of positive selection have been detected in the CC and B30.2 domains, suggesting a long history of interactions with viral pathogens but also endogenous retroviruses [21].
Among the several members of the TRIM family with antiviral activity, we have focused this article on TRIM22 as it targets multiple viruses by exploiting different mechanisms of inhibition. As the TRIM22 N-terminus domain is endowed with E3 ubiquitin ligase activity, poly-ubiquitination of viral proteins leads to their proteasome degradation, whereas the CC domain is engaged in more complex protein–protein interactions with less defined mechanism(s) of viral restriction [22][23].
2. TRIM22 Expression and Protein Localisation
TRIM22, also known as Stimulated Trans-Acting Factor of 50 kDa (Staf50), was first discovered in a cDNA library screening of IFN-α/β-treated Daudi B cells as a gene that was transcriptionally upregulated [24]. TRIM22 is expressed in peripheral blood lymphocytes (PBMC) in response to IFN-α stimulation [24] and constitutively expressed in several human tissues, where it is highly upregulated in response to both type I and type II IFNs [25]. Indeed, the expression of many other TRIM family members is induced by type I and type II IFNs in PBMC [26], suggesting that TRIM proteins represent important mediators of the antiviral response. The 5′ flanking region of the TRIM22 gene contains two regions matching the consensus sequence for an IFN-stimulating response element (ISRE), which are capable of binding IFN regulatory factor 1 (IRF-1) and are important for sensing the stimulation by type I and II IFNs, as well as for basal TRIM22 expression [27]. In addition to IFNs, TRIM22 expression is also modulated in response to several viruses and viral antigens [25]. For example, it is upregulated after infection of rubella virus and Epstein–Barr virus (EBV), but it is downregulated during infection with human papillomavirus type 31 [28], or by the hepatitis B virus (HBV) X protein, thereby allowing HBV to evade the host immune response [29].
The antiviral functions of TRIM22 are also dependent on its subcellular localization as it has been reported to be present both in the cytoplasm [10][30] and in the nucleus [31][32]. This distinct localization has mostly been studied in in vitro systems of ectopic expression. TRIM22 coupled to the green fluorescence protein (GFP) was localized in cytoplasmic bodies in U2OS cells [10], whereas another study reported a diffuse accumulation of the TRIM22-GFP fusion protein surrounding the nucleus of COS-7 cells [30]. A similar localization was observed in HeLa cells expressing endogenous TRIM22 [30]. In contrast, a c-myc-tagged TRIM22 expressed in human PBMC was localized exclusively in the nucleus [33]. Nevertheless, the nuclear expression of TRIM22 is dependent on the B30.2 domain [34], although it has been reported that both a deletion mutant of the RING domain and a cysteine mutant in position 15 of the RING domain disrupt its nuclear localization in HepG2 cells [31]. Furthermore, endogenous expression of TRIM22 has been selectively reported in the nucleus of HeLa cells and U937 cells [35]. The nuclear expression is characterized by the formation of nuclear bodies (NB) similar to TRIM19/PML NB, another member of the C-IV family with antiviral functions [36]. Indeed, TRIM19/PML NB are complex aggregates of proteins that not only include TRIM22 but also the transcription factors class II transactivator (CIITA) and specificity protein-1 (Sp1), as well as Cyclin T1 (CyT1) [37]. These NB favor chromatinization and silencing of viral genomes [38], as in the case of HIV-1 that persists in latently infected cells [39][40].
In the next paragraphs, we will discuss the role of TRIM22 as an antiviral protein against specific viruses.
3. HIV-1
3.1. Life Cycle
Human immunodeficiency virus type 1 (HIV-1) is a member of the lentivirus genus of the retroviridae family that causes a lethal condition known as AIDS (acquired immunodeficiency syndrome) in humans by infecting CD4+ T lymphocytes, causing their depletion and profound immunodeficiency, leading to opportunistic infections and cancer [41]; in addition to CD4+ T lymphocytes, HIV-1 also infects mononuclear phagocytes that are not depleted. After infection, the viral RNA genome is retrotranscribed into DNA that is then integrated as proviral DNA in the host genome [42]. The provirus is actively transcribed during a productive infection by the combined action of the viral protein Tat and of the cellular transcription machinery [43]. Tat is a virus-encoded transcriptional transactivator that binds to the RNA secondary structure of the transactivation region (TAR) of the 5′ long terminal repeat (LTR) (+1 to +59) [44][45] (Figure 1A). Once Tat is bound to the TAR RNA, it recruits a protein complex named positive transcription elongation factor b (p-TEFb) aimed to elongate the viral transcripts. p-TEFb is formed by the regulatory subunit cyclin T1 (CyT1) and the kinase subunit cyclin-dependent kinase 9 (CDK9) that phosphorylate the RNA polymerase II (Pol II) to increase its processivity. However, Tat elongation activity requires a basal transcription that is under the control of the upstream regulatory sequences, namely, three specificity protein 1 (Sp1) and two nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) binding sites that respond to pro-inflammatory signals [46]. The lack of NF-kB and Sp1 binding to the promoter, or the lack of recruitment of negative transcription factors to their DNA binding sites maintains a state of proviral latency [47]. In this regard, latently infected cells (mostly CD4+ T cells) are considered the main obstacle to virus eradication in that they are not affected by combination antiretroviral therapy (cART) [48][49]. The accomplishment of a full HIV-1 life cycle is essential for viral spreading, and it is counteracted by numerous host determinants collectively defined as restriction factors that are constitutively expressed prior to infection and/or are rapidly induced upon pathogen exposure [50]. Among these, other members of the TRIM family have been shown to play a significant role in preventing or containing HIV-1 replication, including TRIM5α [51], TRIM11 [52], TRIM28 [53], TRIM33 [54], TRIM34 [55] and TRIM37 [56].
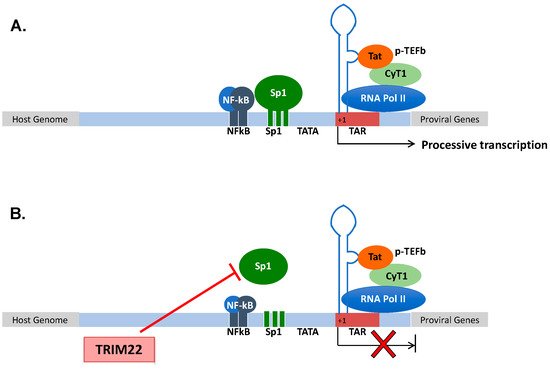
Figure 1. (A) Structural organization of the HIV-1 promoter. HIV-1 transcription starts at the promoter region in the 5′ LTR. Processive HIV-1 transcription is driven by the Tat protein that recruits the p-TEFb complex to the TAR RNA. pTEFb promotes the phosphorylation of the RNA Pol II, enabling the elongation of the viral transcripts. Upstream of the initiation of transcription (+1), three Sp1 and two NF-kB binding sites control the levels of basal transcription and response to inflammatory signals, respectively. (B) TRIM22 inhibits HIV-1 basal transcription by preventing Sp1 binding to the HIV-1 promoter, thus contributing to the maintenance of latent HIV-1 infection.
3.2. TRIM22 Restriction of HIV-1
Since its discovery in 1995, TRIM22 has been characterized for its capacity to impair HIV-1 transcription [24]. Then, TRIM22 was demonstrated to inhibit HIV-1 replication in promonocytic cell lines, and in primary human monocyte-derived macrophages (MDM) [35][57]. Of interest, TRIM22 was shown to inhibit the basal activity of the HIV-1 promoter while not interfering with either the Tat-dependent or NF-kB-mediated upregulation of viral transcription, although it inhibited HIV-1 LTR-mediated gene expression induced by phorbol esters and ionomycin [35]. More recently, TRIM22 was shown to specifically interfere with Sp1-dependent transcription (Figure 1B). Sp1 is a zinc finger transcription factor constitutively expressed in many cell types that binds to GC-rich motifs present in many promoters, and it is involved in many cellular processes, including cell differentiation [58][59], cell growth [60], apoptosis [61], DNA damage response and chromatin remodeling [62].
Although TRIM22 (as with all the other TRIM proteins) does not bind directly to DNA sequences, it prevented the binding of Sp1 to its consensus sites in the HIV-1 LTR, as demonstrated by chromatin immunoprecipitation analysis [63] (Figure 1B).
Recent studies have demonstrated that TRIM22 plays a role in the maintenance of HIV latency in a Tat-independent context, highlighting the effect of TRIM22 on the LTR promoter region, and suggesting a contribution of this protein to the epigenetic silencing of the provirus [64]. More recently, another ISG, namely, interferon-γ inducible protein 16 (IFI16), was shown to restrict HIV-1 by sequestering the transcription factor Sp1, thereby inhibiting viral gene expression [65].
While TRIM22 E3 ubiquitin ligase activity was shown to be required for its interference with the release of HIV-1 particles, likely by interfering with post-translational modifications of HIV-1 Gag proteins [66], it was not required for TRIM22 inhibition of HIV-1 transcription. Therefore, the precise mechanism of TRIM22 interference with HIV-1 transcription is still partially unidentified [35]. TRIM22 did not cause the downregulation of Sp1 expression; nonetheless, TRIM22 inhibited the binding of Sp1 to the HIV-1 promoter, suggesting that a protein complex formed by TRIM22 and other cellular proteins could sequester Sp1, an interpretation supported by the observation that the CC domain of TRIM proteins mediates protein–protein interactions [11]. In this regard, it is worth noting the identification of two single-nucleotide missense polymorphisms (SNP) in the CC domain associated with a loss of inhibition of HIV-1 transcription and HIV-1 disease severity [67]. These two SNPs were discovered by comparing a TRIM22 sequence (as published in GenBank: NM_006074.4) with that of cell clones of the human promocytic cell line U937 that are either non-permissive (“Minus clones”) or permissive (“Plus clones”) to HIV-1 replication [68]. The two SNPs cause an A-to-G transition of SNP rs7935564 with an asparagine-to-aspartic acid substitution in position 155 (Asn155Asp, SNP1), whereas a C-to-G transversion of SNP rs1063303 causes a substitution of a threonine with arginine in position 242 (Thr242Arg, SNP2). Indeed, these two missense mutations affected HIV-1 replication in vitro as PBMC from individuals with the Asn155 and Thr242 haplotypes replicated HIV-1 less efficiently than PBMC with the other mutations. These results were consistent with the ability of TRIM22 to inhibit HIV-1 transcription in vitro. Conversely, the SNP1G variant alone was significantly more frequent in a cohort of HIV-1-infected individuals with advanced disease in comparison to long-term non-progressors (LTNP) or normal progressors [67].
Overall, these results unveiled a role of TRIM22 as a silencer of basal HIV-1 transcription, favoring the maintenance of a state of proviral latency.
4. Influenza A Virus (IAV)
4.1. IAV Infection
Influenza viruses are single-stranded, negative-sense, enveloped RNA viruses of the orthomixoviridae family with a segmented genome composed of eight independent RNA fragments, each one encoding for structural and non-structural proteins. According to the antigenic differences between the nucleoprotein (NP) and matrix (M) protein, influenza viruses can be classified into three types, namely, A, B and C. Although all three types of influenza viruses can naturally infect humans, only the type A virus has a wide range of animal host species, including birds, swine, horses and other mammals [69], whereas the identification of influenza B and C viruses in animal hosts is sporadic [70][71].
IAVs have been extensively studied due to their ability to cause highly contagious diseases in humans and animals (such as poultry, swine and horses), with potentially fatal outcomes [69]. Their intrinsic nature is to continuously change the antigenicity by accumulating point mutations on the surface glycoproteins to escape the existing immunity established by previous infection or vaccination (so-called “antigenic drift”) [69][72]. Furthermore, they cause pandemics by the so-called “antigenic shift”, during which new antigenic subtypes are introduced, by segment reassortment, into an immunologically naïve host population. Further adaptations occur to facilitate transmission in the new host species [73]. Although many global pandemics and major epidemics have occurred at regular intervals during human history [74], during the last century, however, four pandemics have been documented in 1918, 1957, 1968 and 2009 [75]. Then, due to the replicating nature of influenza viruses and the pressure of the immune response, the pandemic viruses progressively evolve into seasonal viruses that acquire mutations to escape the immune response elicited in the previous year [76]. These antigenic changes require an annual update of the seasonal vaccine composition [77]. Interestingly, every 38–40 years, a replacement of the normally circulating seasonal virus with a completely new virus occurs that is not recognized by memory B and T lymphocytes and, thus, causes a pandemic, as most of the population is immunologically naïve [78].
Influenza virus infections induce both innate and adaptive host immune responses, which ultimately result in the abortion of virus replication [79]. Innate immunity and adaptive immunity profoundly differ from each other in terms of responsiveness, specificity and functionality. Innate immunity is the first line of defense against IAV that is specialized in controlling primary infection and induces the adaptive response through the production of co-stimulatory molecules, such as type I IFN, that exhibit antiviral, anti-proliferative and immunomodulatory functions [80]. Thus, antibody-mediated immunity and cellular-mediated immunity become activated and completely neutralize the virus.
4.2. Mechanism of IAV Restriction by TRIM22
IAV induces type I IFNs and ISGs with an antiviral function [81][82][83]. Among these, TRIM22 restricts seasonal IAVs by interacting with the viral NP. The viral NP is a major structural component of the viral ribonucleoprotein (vRNP), a heterotrimeric complex that is bound to the viral RNA and is responsible for viral transcription and replication [84]. In particular, NP binding to viral RNA is crucial for vRNP activity during the elongation phase of vRNA transcription [85]. NP is required to stabilize nascent RNA, which would otherwise be degraded by host cell nucleases. TRIM22 binding to NP promotes its downregulation through ubiquitination and degradation in a proteosome-dependent manner [86]. The TRIM22 RING domain with its E3 ubiquitin ligase activity catalyzes the ligation of previously activated ubiquitin to the lysine residues of the NP [87].
4.3. TRIM22 and IAV Evolution
A wide range of proteomic and genome-wide RNAi-based screens have been used to identify host factors that are partners of NPs and RNPs in viral replication, as reviewed in [88]. However, few factors have been extensively characterized. TRIM22 has the peculiarity of being able to restrict seasonal, but not pandemic, influenza virus replication in vitro [89]. Despite the fact that the NP is a highly conserved protein, differently from the hemagglutinin protein that mediates entry into cells, and that it is the target of neutralizing antibodies [90], in comparison with seasonal pandemic virus sequences, four lysine (K) mutations were identified in seasonal viruses, whereas pandemic viruses were endowed with arginine (R) residues (Figure 2).
Figure 2. Evolution from pandemic to seasonal IAV has shaped TRIM22 restriction. Pandemic viruses are resistant to TRIM22 inhibition as their NP is endowed with four arginine (R) residues that progressively mutate into lysine (K) residues, becoming the target of the U3 ubiquitin ligase activity of TRIM22. The transition of R into K is dependent on viral polymerase errors that generate viral quasispecies either characterized by one, two, three or four K residues. However, a bottleneck of transmission favors the emergence of an IAV NP susceptible to TRIM22 restriction. This phenomenon is likely related to the general rule of viral evolution which endows the virus with the ability to become more transmissible and less pathogenic.
These four R-to-K changes progressively accumulated in approximately 90 years of IAV circulation in humans when sequences from the original pandemic 1918 H1N1 virus were compared with those of the following seasonal strains until 2009, when a new pandemic H1N1 virus emerged. The modeling of the atomic NP 3D structure showed that the four lysine residues are exposed to the solvent and therefore are potential targets of TRIM22 ubiquitination [89]. Concerning the other possible roles of the amino acid R-to-K changes, it has been previously reported that none of these residues are involved in the bipartite nuclear localization signal [91], binding to viral RNA [92][93] and viral polymerases [94], but they are mainly correlated with the host specificity of the virus [95]. In this regard, two sites, i.e., 98 and 422, are part of cytotoxic T lymphocyte (CTL) epitopes [96]. As only two of the four R-to-K variations are likely the result of CTL escape, other selective forces must contribute to the NP variation.
Of relevance is the potential role of adaptive mutations in the IAV animal host that can render viruses resistant to human restriction factors and, thus, have the advantage of being transmitted to humans. In this regard, human myxovirus resistance A (MxA) has been described as a potent restriction factor of avian IAVs [97]; however, the 1918 and 2009 pandemic H1N1 viruses have acquired a cluster of mutations in the NP that inactivates MxA restriction [98]. Mutations conferring MxA resistance are absent in avian IAVs; however, these mutations have been acquired in avian-derived viruses circulating in swine [99]. As pandemic strains are also resistant to TRIM22 restriction, NP adaptation in the swine host could also explain their lack of susceptibility to TRIM22 restriction. However, during IAV evolution in humans, TRIM22 acquires the ability to interact with the NP and adaptive mutations in the NP that render IAVs sensitive to TRIM22 restriction. Indeed, TRIM22 directly interacted with the NP of susceptible IAV strains both in a cotransfection system and during in vitro infection, and this interaction was followed by TRIM22-mediated downregulation and ubiquitination of the viral protein [86]. In contrast, the 2009 pandemic virus and the viral strains that are resistant to TRIM22 activity were unable to interact with TRIM22. Experiments based on the mini-replicon genome system demonstrated that the four NP R-to-K mutations are the main determinants of TRIM22 sensitivity [89].
In order to elucidate the mechanisms that IAV has adopted to escape restriction factors, Juan Ortin’s laboratory demonstrated that, in the absence of the selection pressure exerted by IFNs, serial passages of IAV promoted the introduction of mutations that allowed the virus to increase replication fitness [100]. However, in the absence of any constraint such as that of IAV cultivation in eggs or cell cultures, many of the adaptative mutations acquired during viral passages were purged from the viral population during or shortly after infection, as demonstrated in a human challenge study [101]. In the presence of selection pressure and bottleneck of transmission, IAV may acquire adaptative mutations that could lead to increased susceptibility to restriction factors, including TRIM22, thereby resulting in a less efficient viral replication.
In conclusion, TRIM22 is an IFN-dependent restriction factor of human-adapted IAV, whereas it does not function as a barrier for pandemic viral strains. During replication in animal hosts, the pandemic strains undergo a number of amino acid changes in the NP that render them resistant to TRIM22 restriction and favor their transmission and human-to-human spread. Overall, the genetic variations in the NP gene will be useful for monitoring the viruses and preparing effective prevention and control strategies for potential pandemic influenza outbreaks.
References
- Cao, X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat. Rev. Immunol. 2016, 16, 35–50.
- Shows, T.B.; Sakaguchi, A.Y.; Naylor, S.L.; Goedell, D.V.; Lawn, R.M. Clustering of leukocyte and fibroblast interferon genes of human chromosome 9. Science 1982, 218, 373–374.
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32.
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514.
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485.
- van Gent, M.; Sparrer, K.M.J.; Gack, M.U. TRIM Proteins and Their Roles in Antiviral Host Defenses. Annu. Rev. Virol. 2018, 5, 385–405.
- van Tol, S.; Hage, A.; Giraldo, M.I.; Bharaj, P.; Rajsbaum, R. The TRIMendous Role of TRIMs in Virus-Host Interactions. Vaccines 2017, 5, 23.
- Reymond, A.; Meroni, G.; Fantozzi, A.; Merla, G.; Cairo, S.; Luzi, L.; Riganelli, D.; Zanaria, E.; Messali, S.; Cainarca, S.; et al. The tripartite motif family identifies cell compartments. EMBO J. 2001, 20, 2140–2151.
- Ikeda, K.; Inoue, S. TRIM proteins as RING finger E3 ubiquitin ligases. Adv. Exp. Med. Biol. 2012, 770, 27–37.
- Meroni, G.; Diez-Roux, G. TRIM/RBCC, a novel class of ‘single protein RING finger’ E3 ubiquitin ligases. Bioessays 2005, 27, 1147–1157.
- Sanchez, J.G.; Okreglicka, K.; Chandrasekaran, V.; Welker, J.M.; Sundquist, W.I.; Pornillos, O. The tripartite motif coiled-coil is an elongated antiparallel hairpin dimer. Proc. Natl. Acad. Sci. USA 2014, 111, 2494–2499.
- Rhodes, D.A.; de Bono, B.; Trowsdale, J. Relationship between SPRY and B30.2 protein domains. Evolution of a component of immune defence? Immunology 2005, 116, 411–417.
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C., 3rd. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860.
- James, L.C.; Keeble, A.H.; Khan, Z.; Rhodes, D.A.; Trowsdale, J. Structural basis for PRYSPRY-mediated tripartite motif (TRIM) protein function. Proc. Natl. Acad. Sci. USA 2007, 104, 6200–6205.
- Short, K.M.; Cox, T.C. Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. J. Biol. Chem. 2006, 281, 8970–8980.
- Vernet, C.; Boretto, J.; Mattei, M.G.; Takahashi, M.; Jack, L.J.; Mather, I.H.; Rouquier, S.; Pontarotti, P. Evolutionary study of multigenic families mapping close to the human MHC class I region. J. Mol. Evol. 1993, 37, 600–612.
- Henry, J.; Mather, I.H.; McDermott, M.F.; Pontarotti, P. B30.2-like domain proteins: Update and new insights into a rapidly expanding family of proteins. Mol. Biol. Evol. 1998, 15, 1696–1705.
- Ponting, C.; Schultz, J.; Bork, P. SPRY domains in ryanodine receptors (Ca(2+)-release channels). Trends Biochem. Sci. 1997, 22, 193–194.
- D’Cruz, A.A.; Babon, J.J.; Norton, R.S.; Nicola, N.A.; Nicholson, S.E. Structure and function of the SPRY/B30.2 domain proteins involved in innate immunity. Protein Sci. 2013, 22, 1–10.
- Stremlau, M.; Perron, M.; Welikala, S.; Sodroski, J. Species-specific variation in the B30.2(SPRY) domain of TRIM5alpha determines the potency of human immunodeficiency virus restriction. J. Virol. 2005, 79, 3139–3145.
- Sawyer, S.L.; Emerman, M.; Malik, H.S. Discordant evolution of the adjacent antiretroviral genes TRIM22 and TRIM5 in mammals. PLoS Pathog. 2007, 3, e197.
- Lian, Q.; Sun, B. Interferons command Trim22 to fight against viruses. Cell. Mol. Immunol. 2017, 14, 794–796.
- Vicenzi, E.; Poli, G. The interferon-stimulated gene TRIM22: A double-edged sword in HIV-1 infection. Cytokine Growth Factor Rev. 2018, 40, 40–47.
- Tissot, C.; Mechti, N. Molecular cloning of a new interferon-induced factor that represses human immunodeficiency virus type 1 long terminal repeat expression. J. Biol. Chem. 1995, 270, 14891–14898.
- Hattlmann, C.J.; Kelly, J.N.; Barr, S.D. TRIM22: A Diverse and Dynamic Antiviral Protein. Mol. Biol. Int. 2012, 2012, 153415.
- Carthagena, L.; Bergamaschi, A.; Luna, J.M.; David, A.; Uchil, P.D.; Margottin-Goguet, F.; Mothes, W.; Hazan, U.; Transy, C.; Pancino, G.; et al. Human TRIM gene expression in response to interferons. PLoS ONE 2009, 4, e4894.
- Gao, B.; Wang, Y.; Xu, W.; Duan, Z.; Xiong, S. A 5’ extended IFN-stimulating response element is crucial for IFN-gamma-induced tripartite motif 22 expression via interaction with IFN regulatory factor-1. J. Immunol. 2010, 185, 2314–2323.
- Chang, Y.E.; Laimins, L.A. Microarray analysis identifies interferon-inducible genes and Stat-1 as major transcriptional targets of human papillomavirus type 31. J. Virol. 2000, 74, 4174–4182.
- Lim, K.H.; Park, E.S.; Kim, D.H.; Cho, K.C.; Kim, K.P.; Park, Y.K.; Ahn, S.H.; Park, S.H.; Kim, K.H.; Kim, C.W.; et al. Suppression of interferon-mediated anti-HBV response by single CpG methylation in the 5’-UTR of TRIM22. Gut 2018, 67, 166–178.
- Herr, A.M.; Dressel, R.; Walter, L. Different subcellular localisations of TRIM22 suggest species-specific function. Immunogenetics 2009, 61, 271–280.
- Gao, B.; Duan, Z.; Xu, W.; Xiong, S. Tripartite motif-containing 22 inhibits the activity of hepatitis B virus core promoter, which is dependent on nuclear-located RING domain. Hepatology 2009, 50, 424–433.
- Sivaramakrishnan, G.; Sun, Y.; Tan, S.K.; Lin, V.C. Dynamic localization of tripartite motif-containing 22 in nuclear and nucleolar bodies. Exp. Cell Res. 2009, 315, 1521–1532.
- Duan, Z.; Gao, B.; Xu, W.; Xiong, S. Identification of TRIM22 as a RING finger E3 ubiquitin ligase. Biochem. Biophys. Res. Commun. 2008, 374, 502–506.
- Sivaramakrishnan, G.; Sun, Y.; Rajmohan, R.; Lin, V.C. B30.2/SPRY domain in tripartite motif-containing 22 is essential for the formation of distinct nuclear bodies. FEBS Lett. 2009, 583, 2093–2099.
- Kajaste-Rudnitski, A.; Marelli, S.S.; Pultrone, C.; Pertel, T.; Uchil, P.D.; Mechti, N.; Mothes, W.; Poli, G.; Luban, J.; Vicenzi, E. TRIM22 inhibits HIV-1 transcription independently of its E3 ubiquitin ligase activity, Tat, and NF-kappaB-responsive long terminal repeat elements. J. Virol. 2011, 85, 5183–5196.
- Everett, R.D.; Chelbi-Alix, M.K. PML and PML nuclear bodies: Implications in antiviral defence. Biochimie 2007, 89, 819–830.
- Forlani, G.; Accolla, R.S. Tripartite Motif 22 and Class II Transactivator Restriction Factors: Unveiling Their Concerted Action against Retroviruses. Front. Immunol. 2017, 8, 1362.
- Corpet, A.; Kleijwegt, C.; Roubille, S.; Juillard, F.; Jacquet, K.; Texier, P.; Lomonte, P. PML nuclear bodies and chromatin dynamics: Catch me if you can! Nucleic Acids Res. 2020, 48, 11890–11912.
- Forlani, G.; Tosi, G.; Turrini, F.; Poli, G.; Vicenzi, E.; Accolla, R.S. Tripartite Motif-Containing Protein 22 Interacts with Class II Transactivator and Orchestrates Its Recruitment in Nuclear Bodies Containing TRIM19/PML and Cyclin T1. Front. Immunol. 2017, 8, 564.
- Forlani, G.; Turrini, F.; Poli, G.; Vicenzi, E.; Accolla, R. P-D2 TRIM22 binds to CIITA and sequesters it into nuclear bodies containing TRIM19/PML and Cyclin T1: Implications for HIV-1 infection. JAIDS J. Acquir. Immune Defic. Syndr. 2018, 77, 59.
- Moir, S.; Chun, T.W.; Fauci, A.S. Pathogenic mechanisms of HIV disease. Annu. Rev. Pathol. 2011, 6, 223–248.
- Goodsell, D.S. Illustrations of the HIV life cycle. Curr. Top. Microbiol. Immunol. 2015, 389, 243–252.
- Rice, A.P. The HIV-1 Tat Protein: Mechanism of Action and Target for HIV-1 Cure Strategies. Curr. Pharm. Des. 2017, 23, 4098–4102.
- Berkhout, B.; Silverman, R.H.; Jeang, K.T. Tat trans-activates the human immunodeficiency virus through a nascent RNA target. Cell 1989, 59, 273–282.
- Feng, S.; Holland, E.C. HIV-1 tat trans-activation requires the loop sequence within tar. Nature 1988, 334, 165–167.
- Cullen, B.R. Regulation of HIV-1 gene expression. FASEB J. 1991, 5, 2361–2368.
- Khoury, G.; Darcis, G.; Lee, M.Y.; Bouchat, S.; Van Driessche, B.; Purcell, D.F.J.; Van Lint, C. The Molecular Biology of HIV Latency. Adv. Exp. Med. Biol. 2018, 1075, 187–212.
- Dahabieh, M.S.; Battivelli, E.; Verdin, E. Understanding HIV latency: The road to an HIV cure. Annu. Rev. Med. 2015, 66, 407–421.
- Churchill, M.J.; Deeks, S.G.; Margolis, D.M.; Siliciano, R.F.; Swanstrom, R. HIV reservoirs: What, where and how to target them. Nat. Rev. Microbiol. 2016, 14, 55–60.
- Nchioua, R.; Bosso, M.; Kmiec, D.; Kirchhoff, F. Cellular Factors Targeting HIV-1 Transcription and Viral RNA Transcripts. Viruses 2020, 12, 495.
- Jimenez-Guardeno, J.M.; Apolonia, L.; Betancor, G.; Malim, M.H. Immunoproteasome activation enables human TRIM5alpha restriction of HIV-1. Nat. Microbiol. 2019, 4, 933–940.
- Yuan, T.; Yao, W.; Tokunaga, K.; Yang, R.; Sun, B. An HIV-1 capsid binding protein TRIM11 accelerates viral uncoating. Retrovirology 2016, 13, 72.
- Ma, X.; Yang, T.; Luo, Y.; Wu, L.; Jiang, Y.; Song, Z.; Pan, T.; Liu, B.; Liu, G.; Liu, J.; et al. TRIM28 promotes HIV-1 latency by SUMOylating CDK9 and inhibiting P-TEFb. Elife 2019, 8.
- Ali, H.; Mano, M.; Braga, L.; Naseem, A.; Marini, B.; Vu, D.M.; Collesi, C.; Meroni, G.; Lusic, M.; Giacca, M. Cellular TRIM33 restrains HIV-1 infection by targeting viral integrase for proteasomal degradation. Nat. Commun. 2019, 10, 926.
- Ohainle, M.; Kim, K.; Komurlu Keceli, S.; Felton, A.; Campbell, E.; Luban, J.; Emerman, M. TRIM34 restricts HIV-1 and SIV capsids in a TRIM5alpha-dependent manner. PLoS Pathog. 2020, 16, e1008507.
- Tabah, A.A.; Tardif, K.; Mansky, L.M. Anti-HIV-1 activity of Trim 37. J. Gen. Virol. 2014, 95, 960–967.
- Bouazzaoui, A.; Kreutz, M.; Eisert, V.; Dinauer, N.; Heinzelmann, A.; Hallenberger, S.; Strayle, J.; Walker, R.; Rubsamen-Waigmann, H.; Andreesen, R.; et al. Stimulated trans-acting factor of 50 kDa (Staf50) inhibits HIV-1 replication in human monocyte-derived macrophages. Virology 2006, 356, 79–94.
- Thomas, K.; Wu, J.; Sung, D.Y.; Thompson, W.; Powell, M.; McCarrey, J.; Gibbs, R.; Walker, W. SP1 transcription factors in male germ cell development and differentiation. Mol. Cell Endocrinol. 2007, 270, 1–7.
- Xia, C.P.; Pan, T.; Zhang, N.; Guo, J.R.; Yang, B.W.; Zhang, D.; Li, J.; Xu, K.; Meng, Z.; He, H. Sp1 promotes dental pulp stem cell osteoblastic differentiation through regulating noggin. Mol. Cell Probes 2020, 50, 101504.
- Obad, S.; Brunnstrom, H.; Vallon-Christersson, J.; Borg, A.; Drott, K.; Gullberg, U. Staf50 is a novel p53 target gene conferring reduced clonogenic growth of leukemic U-937 cells. Oncogene 2004, 23, 4050–4059.
- Deniaud, E.; Baguet, J.; Mathieu, A.L.; Pages, G.; Marvel, J.; Leverrier, Y. Overexpression of Sp1 transcription factor induces apoptosis. Oncogene 2006, 25, 7096–7105.
- Malewicz, M.; Perlmann, T. Function of transcription factors at DNA lesions in DNA repair. Exp. Cell Res. 2014, 329, 94–100.
- Turrini, F.; Marelli, S.; Kajaste-Rudnitski, A.; Lusic, M.; Van Lint, C.; Das, A.T.; Harwig, A.; Berkhout, B.; Vicenzi, E. HIV-1 transcriptional silencing caused by TRIM22 inhibition of Sp1 binding to the viral promoter. Retrovirology 2015, 12, 104.
- Turrini, F.; Saliu, F.; Forlani, G.; Das, A.T.; Van Lint, C.; Accolla, R.S.; Berkhout, B.; Poli, G.; Vicenzi, E. Interferon-inducible TRIM22 contributes to maintenance of HIV-1 proviral latency in T cell lines. Virus Res. 2019, 269, 197631.
- Hotter, D.; Bosso, M.; Jonsson, K.L.; Krapp, C.; Sturzel, C.M.; Das, A.; Littwitz-Salomon, E.; Berkhout, B.; Russ, A.; Wittmann, S.; et al. IFI16 Targets the Transcription Factor Sp1 to Suppress HIV-1 Transcription and Latency Reactivation. Cell Host Microbe 2019, 25, 858–872.e13.
- Barr, S.D.; Smiley, J.R.; Bushman, F.D. The interferon response inhibits HIV particle production by induction of TRIM22. PLoS Pathog. 2008, 4, e1000007.
- Ghezzi, S.; Galli, L.; Kajaste-Rudnitski, A.; Turrini, F.; Marelli, S.; Toniolo, D.; Casoli, C.; Riva, A.; Poli, G.; Castagna, A.; et al. Identification of TRIM22 single nucleotide polymorphisms associated with loss of inhibition of HIV-1 transcription and advanced HIV-1 disease. Aids 2013, 27, 2335–2344.
- Franzoso, G.; Biswas, P.; Poli, G.; Carlson, L.M.; Brown, K.D.; Tomita-Yamaguchi, M.; Fauci, A.S.; Siebenlist, U.K. A family of serine proteases expressed exclusively in myelo-monocytic cells specifically processes the nuclear factor-kappa B subunit p65 in vitro and may impair human immunodeficiency virus replication in these cells. J. Exp. Med. 1994, 180, 1445–1456.
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiol. Rev. 1992, 56, 152–179.
- Osterhaus, A.D.; Rimmelzwaan, G.F.; Martina, B.E.; Bestebroer, T.M.; Fouchier, R.A. Influenza B virus in seals. Science 2000, 288, 1051–1053.
- Manuguerra, J.C.; Hannoun, C. Natural infection of dogs by influenza C virus. Res. Virol. 1992, 143, 199–204.
- Cox, N.J.; Subbarao, K. Global epidemiology of influenza: Past and present. Annu. Rev. Med. 2000, 51, 407–421.
- Parrish, C.R.; Kawaoka, Y. The origins of new pandemic viruses: The acquisition of new host ranges by canine parvovirus and influenza A viruses. Ann. Rev. Microbiol. 2005, 59, 553–586.
- Ghendon, Y. Influenza vaccines: A main problem in control of pandemics. Eur. J. Epidemiol. 1994, 10, 485–486.
- Morens, D.M.; Taubenberger, J.K.; Fauci, A.S. The persistent legacy of the 1918 influenza virus. N. Engl. J. Med. 2009, 361, 225–229.
- Yen, H.L.; Webster, R.G. Pandemic influenza as a current threat. Curr. Top. Microbiol. Immunol. 2009, 333, 3–24.
- Nachbagauer, R.; Palese, P. Is a Universal Influenza Virus Vaccine Possible? Annu. Rev. Med. 2020, 71, 315–327.
- Capua, I.; Kajaste-Rudnitski, A.; Bertoli, E.; Vicenzi, E. Pandemic vaccine preparedness--have we left something behind? PLoS Pathog. 2009, 5, e1000482.
- Long, J.S.; Mistry, B.; Haslam, S.M.; Barclay, W.S. Host and viral determinants of influenza A virus species specificity. Nat. Rev. Microbiol. 2019, 17, 67–81.
- Julkunen, I.; Melen, K.; Nyqvist, M.; Pirhonen, J.; Sareneva, T.; Matikainen, S. Inflammatory responses in influenza A virus infection. Vaccine 2000, 19 (Suppl. S1), S32–S37.
- Brass, A.L.; Huang, I.C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009, 139, 1243–1254.
- Karlas, A.; Machuy, N.; Shin, Y.; Pleissner, K.P.; Artarini, A.; Heuer, D.; Becker, D.; Khalil, H.; Ogilvie, L.A.; Hess, S.; et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 2010, 463, 818–822.
- Konig, R.; Stertz, S.; Zhou, Y.; Inoue, A.; Hoffmann, H.H.; Bhattacharyya, S.; Alamares, J.G.; Tscherne, D.M.; Ortigoza, M.B.; Liang, Y.; et al. Human host factors required for influenza virus replication. Nature 2010, 463, 813–817.
- Te Velthuis, A.J.; Fodor, E. Influenza virus RNA polymerase: Insights into the mechanisms of viral RNA synthesis. Nat. Rev. Microbiol. 2016, 14, 479–493.
- Turrell, L.; Lyall, J.W.; Tiley, L.S.; Fodor, E.; Vreede, F.T. The role and assembly mechanism of nucleoprotein in influenza A virus ribonucleoprotein complexes. Nat. Commun. 2013, 4, 1591.
- Di Pietro, A.; Kajaste-Rudnitski, A.; Oteiza, A.; Nicora, L.; Towers, G.J.; Mechti, N.; Vicenzi, E. TRIM22 inhibits influenza A virus infection by targeting the viral nucleoprotein for degradation. J. Virol. 2013, 87, 4523–4533.
- Eldin, P.; Papon, L.; Oteiza, A.; Brocchi, E.; Lawson, T.G.; Mechti, N. TRIM22 E3 ubiquitin ligase activity is required to mediate antiviral activity against encephalomyocarditis virus. J. Gen. Virol. 2009, 90, 536–545.
- Watanabe, T.; Kawaoka, Y. Influenza virus-host interactomes as a basis for antiviral drug development. Curr. Opin. Virol. 2015, 14, 71–78.
- Pagani, I.; Di Pietro, A.; Oteiza, A.; Ghitti, M.; Mechti, N.; Naffakh, N.; Vicenzi, E. Mutations Conferring Increased Sensitivity to Tripartite Motif 22 Restriction Accumulated Progressively in the Nucleoprotein of Seasonal Influenza A (H1N1) Viruses between 1918 and 2009. mSphere 2018, 3.
- Pappas, L.; Foglierini, M.; Piccoli, L.; Kallewaard, N.L.; Turrini, F.; Silacci, C.; Fernandez-Rodriguez, B.; Agatic, G.; Giacchetto-Sasselli, I.; Pellicciotta, G.; et al. Rapid development of broadly influenza neutralizing antibodies through redundant mutations. Nature 2014, 516, 418–422.
- Ozawa, M.; Fujii, K.; Muramoto, Y.; Yamada, S.; Yamayoshi, S.; Takada, A.; Goto, H.; Horimoto, T.; Kawaoka, Y. Contributions of two nuclear localization signals of influenza A virus nucleoprotein to viral replication. J. Virol. 2007, 81, 30–41.
- Ye, Q.; Krug, R.M.; Tao, Y.J. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 2006, 444, 1078–1082.
- Chenavas, S.; Estrozi, L.F.; Slama-Schwok, A.; Delmas, B.; Di Primo, C.; Baudin, F.; Li, X.; Crepin, T.; Ruigrok, R.W. Monomeric nucleoprotein of influenza A virus. PLoS Pathog. 2013, 9, e1003275.
- Marklund, J.K.; Ye, Q.; Dong, J.; Tao, Y.J.; Krug, R.M. Sequence in the influenza A virus nucleoprotein required for viral polymerase binding and RNA synthesis. J. Virol. 2012, 86, 7292–7297.
- Thippamom, N.; Sreta, D.; Kitikoon, P.; Thanawongnuwech, R.; Poovorawan, Y.; Theamboonlers, A.; Suwannakarn, K.; Parchariyanon, S.; Damrongwatanapokin, S.; Amonsin, A. Genetic variations of nucleoprotein gene of influenza A viruses isolated from swine in Thailand. Virol. J. 2010, 7, 185.
- Boon, A.C.; de Mutsert, G.; Graus, Y.M.; Fouchier, R.A.; Sintnicolaas, K.; Osterhaus, A.D.; Rimmelzwaan, G.F. Sequence variation in a newly identified HLA-B35-restricted epitope in the influenza A virus nucleoprotein associated with escape from cytotoxic T lymphocytes. J. Virol. 2002, 76, 2567–2572.
- Haller, O.; Staeheli, P.; Schwemmle, M.; Kochs, G. Mx GTPases: Dynamin-like antiviral machines of innate immunity. Trends Microbiol. 2015, 23, 154–163.
- Manz, B.; Dornfeld, D.; Gotz, V.; Zell, R.; Zimmermann, P.; Haller, O.; Kochs, G.; Schwemmle, M. Pandemic influenza A viruses escape from restriction by human MxA through adaptive mutations in the nucleoprotein. PLoS Pathog. 2013, 9, e1003279.
- Dornfeld, D.; Petric, P.P.; Hassan, E.; Zell, R.; Schwemmle, M. Eurasian Avian-Like Swine Influenza A Viruses Escape Human MxA Restriction through Distinct Mutations in Their Nucleoprotein. J. Virol. 2019, 93.
- Perez-Cidoncha, M.; Killip, M.J.; Oliveros, J.C.; Asensio, V.J.; Fernandez, Y.; Bengoechea, J.A.; Randall, R.E.; Ortin, J. An unbiased genetic screen reveals the polygenic nature of the influenza virus anti-interferon response. J. Virol. 2014, 88, 4632–4646.
- Sobel Leonard, A.; McClain, M.T.; Smith, G.J.; Wentworth, D.E.; Halpin, R.A.; Lin, X.; Ransier, A.; Stockwell, T.B.; Das, S.R.; Gilbert, A.S.; et al. Deep Sequencing of Influenza A Virus from a Human Challenge Study Reveals a Selective Bottleneck and Only Limited Intrahost Genetic Diversification. J. Virol. 2016, 90, 11247–11258.
More
Information
Subjects:
Cell Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
841
Revisions:
2 times
(View History)
Update Date:
29 Jul 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No