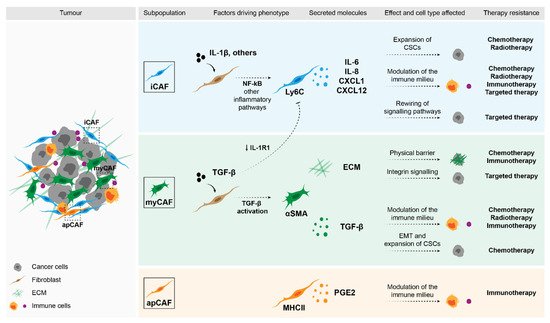
An important way through which fibroblasts modulate the response of tumours to cytotoxic agents, such as chemotherapy and radiotherapy, is via their interaction with CSCs. These cells are at the top of the tumour hierarchy since, in addition to their self-renewal capacity, they are also able to differentiate into committed tumour cells and, consequently, repopulate the whole tumour
[76]. CSCs have been shown to be intrinsically more resistant to these agents, not only due to an enhanced DNA damage repair mechanisms or a higher expression of ATP-binding cassette (ABC) transporters
[77], but also due to their cycling state. CSCs are often in a dormant, non-proliferative state
[78] that makes them relatively refractory to treatments that target an active proliferative state in cells. A mechanism through which fibroblasts have been shown to drive the expansion of the CSCs fraction in tumours was described by Su and colleagues
[60]. The authors showed that a subpopulation of fibroblasts characterised by the expression of two markers, CD10 and GPR77, promoted chemoresistance by driving the survival of CSCs in both breast and lung cancer models. Interestingly, they showed that this subpopulation of fibroblasts is present in the tumours prior to any treatment, and that it becomes enriched upon therapy. CD10
+GPR77
+ fibroblasts were characterised by the activation of the NF-κB pathway, and their secretion of IL-6 as well as IL-8 was essential to drive resistance to chemotherapy. Based on more recent knowledge on the different subsets of fibroblasts, it could be hypothesised that these GPR77- and CD10-positive fibroblasts form a subcluster of the iCAFs. Kanzaki
[79] has shown that, in breast cancer, the expression of CD10 and GPR77 is restricted to one of the clusters identified in a study from Bartoschek and colleagues
[35]. Unfortunately, in this early study, no inflammatory CAF cluster was defined and the parallel is thus hard to establish. Nevertheless, the study from Su et al. further highlights the importance of uncovering fibroblast heterogeneity in the context of cancer. Moreover, it supports the significance of regarding the fibroblast dynamics during therapy. Several other studies have described mechanisms through which fibroblasts can support and expand tumour-initiating cells. Boelens et al. uncovered a paracrine crosstalk between cancer cells and stromal fibroblasts in which RIG-I and NOTCH signalling cooperate to drive the expansion of therapy resistant CSCs
[59]. Briefly, they showed that exosomes secreted by stromal fibroblasts activate the anti-viral machinery in cancer cells via STAT1, which, in turn, drives the expression of interferon-stimulated genes (ISGs). This signature had previously been identified as a gene signature for radiotherapy and chemotherapy resistance
[80][81] and was termed as interferon-related DNA damage resistance signature (IRDS). By facilitating the expression of NOTCH target genes in a STAT1-dependent manner, the stromal interaction with cancer cells drove the expansion of CSCs and, consequently, resistance to chemotherapy. Further studies have implicated JAK-STAT and NF-κB signalling in CAFs in the support of tumour-initiating cells. In pancreatic cancer, Chan et al. demonstrated that the secretion of ELR
+ chemokines, CXCL1, CXCL2, CXCL5 and CXCL6 by CAFs bind and activate the CXCR2 downstream signalling in cancer cells, driving the expansion of CSCs
[82]. Other pathways that modulate stem-like features in cancer cells have also been described to be activated by fibroblasts, namely Hedgehog
[61] and Wnt signalling
[83][84].
Several of the mechanisms described above seem to be mediated by iCAFs. Nevertheless, the myCAFs and ECM they synthesise can also strongly affect the way tumours respond to therapy. For example, a desmoplastic tumour can create a physical barrier that will prevent the exposure of cancer cells to the drug
[85][86]. Moreover, the activation of integrin signalling in cancer cells as a result of a dense ECM, can promote the survival of malignant cells
[87][88]. Modulation of the ECM by fibroblast-derived Anexin A6-loaded extracellular vesicles (EVs) also resulted in integrinβ1-FAK-YAP activation and drove chemoresistance in gastric cancer
[89]. The secretion of immunosuppressive factors such as TGF-β by myCAFs can inhibit ICD and, consequently, impair the effect of radiation and chemotherapy in tumours
[90]. In addition to its effect in immune cells, TGF-β can also modulate the processes of epithelial-to-mesenchymal transition (EMT) in cancer cells as well as regulate the expansion of CSCs
[91].
 +1 point
+1 point