1. Introduction
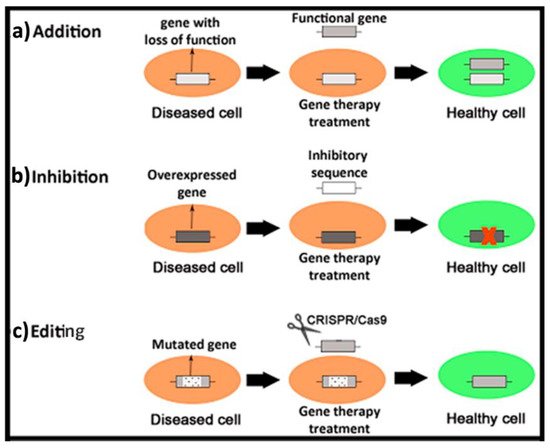
The main concept of gene therapy is quite simple and overall relies on the delivery of exogenous genetic material into target cells to modulate the expression of an altered genome. Basically, three different approaches can be identified (
Figure 1). In the case of
gene addition therapy, a “healthy” copy of the gene is administered to recover the functionality of the affected cells. This strategy can be suitable to face diseases caused by mutations with loss of function
[1]. For instance, the autosomal recessive cystic fibrosis disease caused by a deletion of the phenylalanine at the position 508 of the CFTR (cystic fibrosis transmembrane conductance regulator) protein
[2]. However, in the case of a mutation that overexpresses genes, the aim is to administer an inhibitory sequence to knock out the expression of the mutated gene
[3]. This strategy is referred to as
gene inhibition therapy and can be applied, for instance, to face autosomal dominant retinitis pigmentosa secondary to specific mutations in the pre-mRNA splicing-factor gene PRPF31
[4]. The third approach, named as
genome editing, incorporates specific genome editing tools to repair mutations in the genome with gain or loss of function
[5]. This strategy has been successfully used in combination with iPSC technologies to combat human β-thalassemia disease in mice
[6].
Figure 1. Brief schematic representation of three different genetic material-based approaches to face human diseases. (a) Gene addition therapy. (b) Gene inhibition therapy. (c) Genome editing.
The main characteristics and composition of the genetic cargo strongly depend on the gene therapy approach used. For instance, in the case of the
gene addition approach, which is classically used to face genetic disorders that follow an autosomal recessive inheritance pattern, the most common polynucleotide used is referred to as plasmid (pDNA). Such a plasmid is a circular and double-stranded DNA construct, typically between 1.5 and 20 kbs, that drives the transient transgene expression in the nucleus of target cells, encoding the protein of interest
[7]. Typically, a transfection mediated by conventional pDNAs is moderated and only active during 1–2 months. However, smaller versions of a conventional pDNA, known as minicircle DNA (mcDNA, 2–6 kbs) or micro-intronic plasmid (2–4 kbs), can improve transgene expression by 10- to 100-fold and prolong the effect for some years
[8][9]. Another commonly used polynucleotide in the
gene addition approach is the single strand messenger RNA (mRNA). Nowadays, this strategy holds great promise, especially in the vaccine research area, since two vaccines produced by Moderna and Pfizer/BioNTech companies have been recently approved by the European Medicines Agency (EMA) to fight against the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection and the resulting coronavirus disease (COVID-19). As in the case of pDNA, the effect mediated by mRNAs is also transient and the stability in plasma is even lower, around 1 h. The less tight conformation of RNA, which allows an easier access of the degradative enzymes, and also the presence of hydroxyl groups in the main structure, which enhances hydrolyzation of RNAs
[10][11], significantly contribute to decrease the stability. However, the main advantages of RNA-based gene therapy compared to pDNA include a safer profile, since it decreases the risk of mutagenesis and immunogenicity and a more efficient modulation of target gene expression because the place of action of this genetic cargo is in the cytoplasm
[12]. Therefore, there is no need to get access into the nucleus of cells, which is classically considered as one of the main bottlenecks of plasmid-based expression systems
[13].
In contrast to
gene addition strategy, the
inhibition approach can be used when the main goal is to silence the expression of an altered gene that has a gain of function mutation. This scenario is common in genetic disorders that follow an autosomal dominant inheritance pattern
[14]. In this case, the inhibitory sequence can also have a single strand RNA-based structure, such as the microRNA (miRNA), or a double strand RNA structure, such as the small interfering RNA (siRNA). Typically, these structures inhibit the translation of mRNA in the RNA-induced silencing complex (RISC) of the cytoplasm in a transitory way to avoid the expression of the target gene
[15]. Interestingly, other synthetic and smaller single strand RNA/DNA structures, known as antisense oligonucleotides (AON), can also inhibit mRNA translation by a different mechanism of action. In this case, the oligonucleotide sequence interferes with pre- and mRNA in the nucleus or cytoplasm through a complementary hybridization mechanism that enhances specificity but with lower knockdown efficiency
[16].
Apart from
gene addition and
gene inhibition strategies, another approach consists of the permanent correction of the mutated gene with the use of specific genome editing tools such as those developed by the game changer CRISPR/Cas9 technology
[17]. In this case, once the mutated gene is identified, different RNA guides (gRNA), which recognize 20 nucleotides of the mutated allele, can be designed and synthetized to be delivered alongside the Cas9 protein, which will cut the genome 3 nucleotides to the left side in the 5′-3′ direction of the protospacer adjacent motif (PAM) region. In this scenario, a single strand DNA sequence with the corrected mutation, typically 100 nucleotides long, can be supplied as a donor template to be incorporated in the cell genome by the homologous recombination mechanism
[18]. Such CRISPR/Cas9 editing tools can be delivered in different genetic constructors such as pDNA, mRNA or ribonucleoprotein (RNP) complex, acting in different cell places
[19].
Unfortunately, all the previously described genetic cargoes need to overcome both extracellular and intracellular barriers to reach the place of action. In the case of in vitro conditions, which is the simplest scenario, only intracellular barriers need to be considered. However, in the case of in vivo experimentation, the delivery process to the place of action can also be affected by additional extracellular barriers, which strongly depends on the route of administration and the organ to be treated
[20]. To overcome such biological barriers, gene delivery systems are necessary, since genetic cargo by itself, in most of the cases, is not effective. Classically, gene delivery systems are divided into viral and non-viral vectors. Viral vectors are recognized by their high gene delivery efficiency. In fact, viruses have evolved along many years to infect efficiently different kinds of cells with their genetic cargo, and currently, such viruses can be easily modified in the laboratory to deliver the genetic cargo of interest into target cells, reducing their pathogenic effect
[21]. As a result, most of the clinical trials, and the great majority of gene therapy drugs approved for human use by regulatory agencies, are based on viral vectors. Some examples of marketed gene therapy products that use viral vectors include Luxturna, Zolgensma, Oncorine and Imlygic, to name just a few
[22][23]. However, relevant concern still remains in the research community related, over all, to their potential immunogenicity and oncogenic capacity
[24][25]. In addition, previously mentioned approved drugs are highly expensive, mainly due to the intrinsic characteristics of biologic drugs
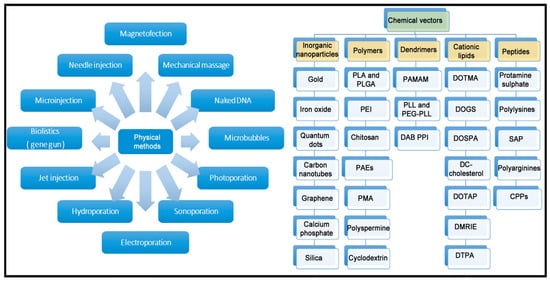
[26]. Therefore, interest in non-viral gene delivery systems has recently gained momentum. A brief schematic representation of both physical and chemical methods for non-viral gene delivery is summarized in
Figure 2.
Figure 2. Overview of main physical and chemical methods of non-viral vectors.
Compared to their viral counterparts, non-viral vectors show some appealing properties such as lower immunogenicity, safer profile and higher genetic cargo packing capacity. In addition, non-viral vectors are cheaper and easier to manufacture and scale up
[27]. Due to these obvious advantages, the gene delivery mediated by non-viral vectors is nowadays considered the cornerstone of modern gene therapy, especially for CRISPR/Cas9 delivery, where non-viral vectors predominate over viral vectors at the preclinical level
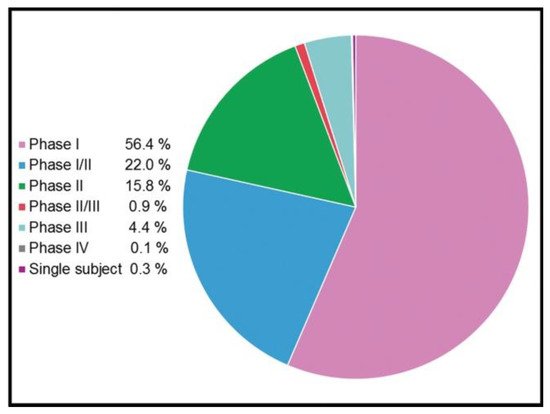
[28]. In any case, although with few exceptions, this strategy has been poorly translated into clinical success. However, some promising clinical trials based on gene therapy treatments, summarized in
Figure 3, are ongoing.
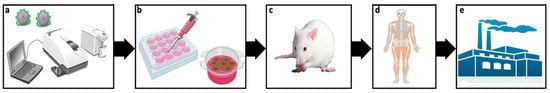
In this review, some critical issues in the way to clinic application of non-viral vectors (Figure 4) and potential strategies to overcome such hurdles have been addressed. More specifically, special attention has been paid to the gene delivery efficiency and biocompatibility of non-viral vectors. Additionally, the duration of the transgene expression, along with the route of administration, the design of experimental conditions and some concerns related to the commercialization process, has also been discussed.
Figure 4. Journey of non-viral vectors from lab to bench. Initially potential non-viral vectors are physicochemically characterized (a) before performing both in vitro (b) an in vivo (c) biological studies. Most promising formulations are further evaluated in clinical trials (d). In case of success, the manufacturing process for commercialization starts (e).
2. Gene Delivery Efficiency
A critical issue that hampers the regular application of non-viral vectors into regular medical practice is their low gene delivery efficiency
[29]. In this sense, viral systems clearly surpass non-viral counterparts, probably due to their continuous evolution along millions of years, which has allowed them to get better access into the genome of the target cell, overcoming both extracellular and intracellular barriers
[30]. Nowadays, most of the commercially available gene therapy-oriented drugs use recombinant viruses modified in the laboratory, such as retroviruses, lentiviruses, adenoviruses or adeno-associated viruses to shuttle their genetic cargo. However, their overall safety concerns related to the biological origin and the low genetic cargo packing capacity, along with the difficulties associated to scaling up their production and high cost of development, have contributed to exploring different gene delivery approaches based on the design of novel non-viral vectors
[31]. Research on this area has quickly captured the attention of the scientific community, and, currently, this strategy represents a safer and more affordable alternative, although the gene delivery efficiency of these systems needs to be improved to reach a regular clinical practice.
Gene delivery efficiency of non-viral vectors is typically evaluated at a preclinical level once such systems have shown appropriate physicochemical and biophysical properties, for instance, in terms of particle size, superficial charge, polydispersity, morphology and capacity to bind and protect genetic material to release it without suffering any degradation, since all these parameters can affect the transfection process
[32]. Initially, and as a proof of concept, gene delivery capacity is evaluated in culture cells and, for that purpose, the expression of different reporter plasmids that encode fluorescent proteins
[33] or enzymes, such as luciferase
[34] or galactosidase
[35], is employed to be quantitatively evaluated by different techniques. In this sense, it is worth mentioning that each reporter plasmid and corresponding assays have different sensitivity and their own metrics
[36]. For example, the reporter plasmid that encodes green fluorescent protein is a good descriptor of the transfection efficiency at a single cell’s level, since results are typically expressed as the percentage of live cells that show green signal by flow cytometry
[37]. However, luciferase expression provides information related to the plasmid expression in a whole population of cells, since the luminescence is normalized by the quantity of proteins in cell lysates.
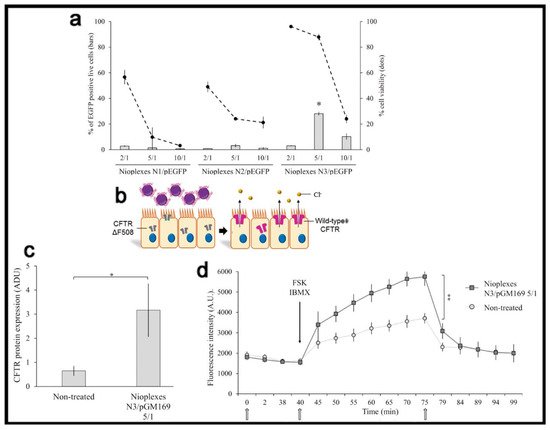
When a therapeutic genetic material, instead of the reporter one, is used in in vitro conditions, it is also important to consider, from a practical point of view, the transfection efficiency value required to reach a therapeutic effect, which highly depends on the particular application and disease. For instance, in the case of cystic fibrosis, an autosomal recessive disorder caused by the dysfunction of the CFTR gene, 28% of living human cystic fibrosis airway epithelial cells (CuFi-1) were transfected with the pEGFP reporter plasmid, using a lipid-based non-viral vector, named as N3
[38]. Such formulation reported a 5-fold increase of CFTR protein expression in transfected versus non-transfected cells with the pGM169 therapeutic plasmid, which led to 1.5-fold increment of the chloride channel functionality, exceeding the value required to get a therapeutic benefit (
Figure 5).
Figure 5. Transfection efficiency and therapeutic effect. (
a) Percentage of live transfected CuFi-1 cells with the pEGFP reporter plasmid. (
b) General scheme of transfection process with pGM169 therapeutic plasmid. (
c) CFTR/β-actin protein expression determined by Western blot. (
d) CFTR chloride channel activity determined by SPQ analysis. Reproduced with permission of
[38].
Such in vitro studies are typically used as a screening methodology to select the non-viral vector candidates that show better performance before conducting in vivo studies, in accordance with the principle of the three Rs (replacement, reduction and refinement of animal labs). This sequential approach is aimed to reduce the number of animals used in in vivo experiments. However, it should also be borne in mind that a direct correlation between in vitro and in vivo results in terms of gene delivery efficiency does not always exist, essentially because experimental conditions in each scenario are quite different
[37]. As a consequence, some readjustments in terms of the composition of the formulation, preparation methods, doses or volumes to be administered need to be performed to succeed in in vivo experiments
[39].
In any case, the transfection efficiency of non-viral vectors is highly related to their cytotoxic effect, which is also a highly cell-dependent process
[40]. Therefore, a suitable balance between the transfection efficiency value required to obtain a therapeutic effect and the cytotoxic effect needs to be acquired for each clinical application to enhance translation of non-viral vectors to the regular medicine practice. Such a toxic effect of non-viral vectors depends on many physicochemical parameters that affect the gene delivery process such as particle size, morphology and zeta potential of complexes
[41]. In addition, the elaboration method, along with the intrinsic properties of the materials used to obtain the different kinds of non-viral vectors, can impact on the final cytotoxic effect, depending, for instance, on the degradation rate or the persistence along the time in organs and tissues
[42]. It should also be kept in mind that the persistence and accumulation of metabolites that come from the degradation of different compounds present in non-viral vector formulations can also induce an inflammatory process and, therefore, cause toxicity. Nevertheless, the cytotoxic effect does not only depend on the non-viral vector’s compounds. The genetic material that is aimed to be delivered can also be toxic, considering, for instance, the bacterial origin of many plasmids that can enhance the induction of undesired immune responses and the secretion of proinflammatory cytokines
[43]. Interestingly, small plasmidic cassettes as mcDNA have been recently developed to mitigate some disadvantages associated with the use of conventional plasmids
[31]. Such mcDNAs contain a minimal expression cassette, where the bacterial backbone DNA has been eliminated, which reduces the unwanted immunogenic responses and enhances the transfection efficiency due to the reduced size of this CpG-free genetic material
[8][44]. The cytotoxic effect of the non-viral vectors can be qualitatively evaluated by different techniques based on microscopy analyses
[45]. However, normally, quantitative analysis of toxicity is assessed by means of a broad spectrum of cell viability/cytotoxicity colorimetric available kits, such as, for instance, CCK8, MTT assay and Alamar Blue
TM, or by mean of flow cytometer analysis
[46][47][48]. In this sense, it is worth mentioning that many chemical compounds that are present in non-viral vector formulations can interfere with the previously described colorimetric assays, providing confusing results. In the case of flow cytometer analysis, fluorescent dyes such as ethidium homodimer-1, propidium iodide or 7-Amino-actinomycin D (7-AAD) are normally used to stain and analyze dead cells, which should be excluded from the final transfection efficiency results
[49].
To reduce the cytotoxic effect of non-viral vector formulations, many natural compounds, such as cholesterol
[50], lycopene
[39] or squalene
[33], can be incorporated into lipid vesicles as “helper” components. In addition, some non-ionic surfactants, such as polysorbate 80, can also reduce the toxic effect of cationic lipids
[51]. Although the use of cationic materials, such as the mentioned cationic lipids or polycationic polymers, facilitates the complexation with the negatively charged nucleic acids for gene delivery as well as cellular internalization, an excess of positive charge can have detrimental effects on cell viability. Hence, other strategies to avoid cationic vectors, and thus cytotoxicity, have been developed for nucleic acid delivery
[52]. In the case of polymeric-based non-viral vector formulations,
stimuli responsive polymers, also knowns as
intelligent polymers, represent an appealing approach to enhance not only biocompatibility of the formulation but also the specificity and the duration of the gene expression
[53]. These particular polymers can modify their biological performance in response to small environmental changes of physicochemical parameters such as pH value, temperature or ionic strength to name just some of the most relevant ones
[54].
3. Duration of Gene Expression
Another main reason that limits the clinical application of non-viral vectors into regular medical practice is the loss of transgene expression over time in clinical trials. A transgene expression can decrease with time because of many causes such as the inactivation of the genetic material by nucleases, the loss of activity by recombination processes, the ineffective distribution into intracellular vesicles, or even the recognition and subsequent silencing of foreign DNA by the host immune system
[55]. In this sense, while the retroviral and lentiviral vectors do integrate into the host cell genome, providing a long-lasting effect
[56][57], the main reason for adeno-associated viruses (AAV) vectors to provide sustained transgene expression is not integration. AAV vectors barely integrate into the genome unlike wild-type AAV. In contrast, an AAV vector genome persists in the host cell nucleus as episomal concatemers that are highly resistant to nucleases. The fact that AAV genomes are diluted over time as the cell undergoes repeated rounds of replication, with the rate of transgene loss dependent on the turnover rate of the transduced cell
[58], is a proof of the occurrence. For instance, commercially available Luxturna drug delivers by means of an AAV type 2 a healthy copy of the RPE65 gene into the subretinal space of patients affected by retinitis pigmentosa and Leber congenital amaurosis. Despite the high cost of the treatment, around $850,000, only one injection is required to complete the treatment, due to the long-lasting effect obtained, in slow dividing cells of the retina. This fact is particularly relevant in the case of invasive routes of administrations, such as intravitreal, subretinal or administrations, into the cerebral cortex after craniotomy. In this scenario, repeated administrations could increase the after-care cost due to additional hospital visits
[59], and, in many cases, jeopardize the acceptance of these aggressive gene delivering routes because of the cumbersome approach and related side effects.
Most of the strategies that have been developed by the research community to enhance the lasting effect of transgene expression are mainly focused on modifications of the genetic material to be delivered rather than on modification on the components of the non-viral vector formulation. For example, in the case of the
gene addition approach, the previously described mcDNA technology not only reduces the cytotoxic effect but also represents a promising approach to prolong the therapeutic effect when this genetic cargo is combined with non-viral vectors
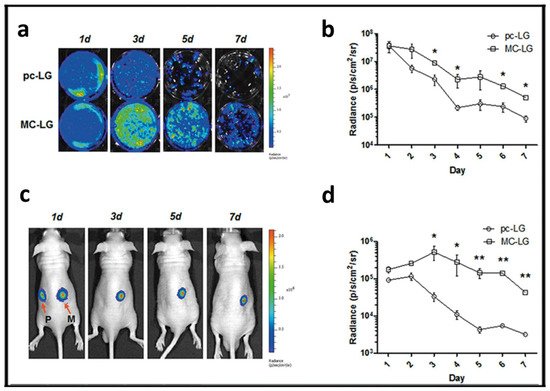
[8]. The lack of bacterial backbone sequences, along with the low content of unmethylated CpG dinucleotides, reduces the activation of nuclear transgene silencing mechanisms, which finally results in a sustained transgene expression effect (
Figure 6,
[60]).
Figure 6. Minicircle approach to transfect mesenchymal stem cells (MSCs). (
a) Bioluminescence images of MSCs transfected with pcDNA3.1-fLuc-2A-EGFP (pc-LG) or McCMV-fLuc- 2A-EGFP (MC-LG). (
b) Quantification of bioluminescence signal emitted by the MSCs transfected with pc-LG (circle) or MC-LG (square). (
c) Bioluminescence images of nude mice subcutaneously injected with MSCs transfected with pc-LG (P) and MC-LG (M) into left and right back. (
d) Quantification of bioluminescence emitted from the injected area of mice. Reproduced with permission of
[60].
In addition, to prolong the effect, the transgenes of interest can be incorporated into the host genome by means of viral integrase or site-specific recombinase enzymes, or by the addition of transposable elements, such as transposons or “jumping” genes
[55]. In any case, the translation into the clinic of these promising approaches to enhance the transgene expression effect is clearly conditioned by relevant safety issues such as the possible induction of insertional mutagenesis in the host cells with permanent consequences. Another approach that can be used to prolong the transgene expression effect consists of the addition of viral DNA sequences that allow plasmid replication outside the chromosomes. However, again, safety concerns can arise due to the viral DNA nature that is associated with immune responses and the risk of oncogenesis. As a safer alternative to the aforementioned viral sequences, mammalian scaffold/matrix attachment regions (S/MARs) can also be incorporated into pDNA to enhance the episomal replication of plasmids
[61]. Episome sequences autonomously replicate plasmids that do not need to be integrated into the host genome to express the transgene, which minimizes the mutagenesis risk
[62]. Episomal replicating plasmids are especially interesting to face tumor cells by gene therapy, where a vertical transfer of the therapeutic plasmids is particularly relevant in fast-dividing malignant cells
[63]. Gene expression can also be prolonged by specific inhibition of gene silencing mechanisms of cells that hampers transgene expression
[64]. Interestingly, artificial transcriptional activators can also be incorporated into therapeutic plasmids with appropriate promoters to modulate gene expression. In the case of the
gene inhibition approach used to silence the expression of an altered gene with inhibitory sequences such as miRNA, siRNA or AON delivered by non-viral vectors, the effect is also transient, which requires repeated administrations
[32]. In this case, a permanent correction of the altered gene can be achieved with the use of CRISPR/Cas9 technology. Such genome editing tools can be delivered by non-viral vectors complexed to different genetic constructs such as pDNA, mRNA or RNP complexes
[19]. In any case, when time is a crucial factor, differences in anatomy, physiology, development and biological phenomena between animal labs and human beings should be also considered to extrapolate experimental results. For instance, it has been estimated that one lived day for rats is comparable to 30 lived days for humans
[65].
4. Administration Route
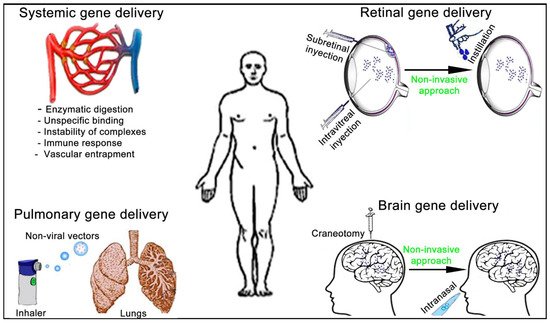
The administration route of non-viral vectors affects not only the previously described gene delivery efficiency and duration of gene expression but also the design of the formulations. To be active at the place of action, non-viral vectors need to overcome different biological barriers that strongly depend on both the administration route and the target organ (Figure 7).
Figure 7. Schematic representation of the main administration routes to face systemic and tissue-specific human diseases by non-viral vectors gene therapy approach.
The main and most studied administration route of non-viral vectors to face disseminated cancer or infectious diseases is the intravenous route
[66]. The idea is to transport the genetic material to as many cancerous and infected cells as possible. However, efficiency of this interesting and ambitious approach is strongly limited by the enzymatic degradation that a genetic material can suffer from the point of entry
[23]. In addition, positively charged non-viral vector complexes can interact by non-specific electrostatic attractions with negatively charged biological compounds such as serum proteins and blood cells, which limit their final performance
[67]. Another concern to be aware of is the possible instability of such complexes in the extracellular biological medium at physiological conditions, where pH value, temperature and ionic stench, among many other factors, can result in the formation of aggregates along the exposure time
[18]. Furthermore, the systemic administration at high volumes of non-viral vector complexes can trigger the host immune responses against some of their components, resulting in an inflammatory response, which can be more pronounced if administrations are repeated
[68]. Finally, it is worth mentioning that endothelial cells of the vascular system constitute a relevant and effective biological barrier. This barrier can limit the size and number of non-viral vector complexes that can pass through it, therefore, reducing the transfection efficiency in the targeted tissues after a systemic administration
[69].
To overcome the previously described systemic barriers, non-viral vectors can be structurally modified. For instance, the addition of positively charged protamine into non-viral vectors protects the genetic material from enzymatic digestion and, therefore, increases the transfection efficiency. This approach has also been successfully used with other lipid formulations such as solid lipid nanoparticles
[70] or liposomes
[71]. Other commonly used strategy to increase the stability of non-viral vectors complexes in biological fluids and reduce the immune response consists in the addition of poly(ethylene glycol) (PEG) chains or other hydrophilic polymers, such as poly(N-vinyl-2-pyrrolidone) (PVP), into the outer surface of complexes
[72]. The neutral PEG chains produce a steric barrier against enzymatic degradation and avoid aggregation of non-viral vector complexes in systemic circulation
[73]. In addition, PEG chains reduce the quantity of cationic lipids required to deliver a genetic material, which enhances biocompatibility of lipid formulations
[74].
Another main route of administration to face devastating diseases that affect the lungs, such as cancer, cystic fibrosis or asthma, is the pulmonary route. In this case, intratracheal intubation can be used for gene delivery of pulmonary disease-oriented treatments. However, the non-invasive nature of the inhalation approach is preferable for clinical applications. Another advantage of this administration route includes the use of small doses, which in turn, reduces side effects by increasing drug concentration at the area of interest. In addition, inhalation can also be used for gene delivery to treat systemic diseases due to the quick absorption on the alveolar region of lungs
[75]. In any case, the effectiveness of the inhalation process depends mainly on the amount of the genetic material that will finally reach the targeted region and the deposition pattern, which is deeply conditioned by the composition of the material to be delivered and by the device used for inhalation
[76]. In this sense, relevant issues such as the characteristics of the drug delivered, the most adequate pattern for its delivery, the design of the device and its effectiveness need to be considered in detail to enhance the effectiveness of this appealing administration route. In the specific case of genetic material-based formulations, the main difficulty to reach the nucleus of target cells is related to the susceptibility of such molecules to be degraded by the hydrodynamic shear forces generated during aerosolization process, which result in a clear decrease of the efficiency compared to in vitro conditions
[77]. Some interesting approaches to protect DNA during the aerosolization process consist of the incorporation of compounds such as bovine serum albumin (BSA), which stabilizes the supercoiled DNA
[78], or the design of efficient aerosol delivery systems for genetic material, such as nebulizers, dry powder inhalers (DPIs), pressurized metered dose inhalers (pMDIs) or mechanical metered dose inhalers (mMDIs)
[79].
 +1 point
+1 point