 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ursula Vogl | + 1648 word(s) | 1648 | 2021-07-06 04:08:46 | | | |
| 2 | Lindsay Dong | Meta information modification | 1648 | 2021-07-27 04:28:51 | | |
Video Upload Options
The cure rate of germ cell tumours (GCTs) has significantly increased from the late 1970s since the introduction of cisplatin-based therapy. The exquisite cisplatin sensitivity has been mainly explained by the over-expression in GCTs of wild-type TP53 protein and the lack of TP53 somatic mutations; however, several other mechanisms seem to be involved, many of which remain still elusive. The findings about the role of TP53 in platinum-sensitivity and resistance, as well as the reported evidence of second cancers (SCs) in GCT patients treated only with surgery, suggesting a spectrum of cancer predisposing syndromes, highlight the need for a deepened understanding of the role of TP53 in GCTs.
1. Introduction
Testicular germ cell tumours (TGCTs) are the most prevalent solid tumours in young men aged 15–35 years, with a rising incidence among Caucasians, although they represent overall 1% of all malignancies in men worldwide [1]. It has been estimated that 23,000 new cases of TGCT in Europe will be diagnosed each year by 2025, with an increase of 24% compared with 2005 [2]. The most reassuring data are that TGCT is one of the most curable solid cancers, with approximately 95% of men surviving at 5 years [3]. This percentage rises to 98–99% for patients diagnosed with clinical stage I (CSI) [4]. The cure rate has significantly increased from the late 1970s since the introduction of cisplatin-based therapy in the [5][6], which to date remains the milestone of germ cell tumours (GCTs) treatment. The exquisite cisplatin sensitivity has been for a long time mainly explained by the over-expression of wild-type TP53 protein and the lack of TP53 somatic mutations; however, several other mechanisms seem to be involved, many of which remain still elusive [7]. Nevertheless, approximately 10–15% of patients will develop a tumour relapse after initial treatment or a refractory disease [8] and non-seminomas, particularly those of extra-gonadal origin, are associated with a lower rate of platinum responses [9][10]. In recent years, these issues have boosted the question of platinum resistance in GCTs, which represents an ongoing unmet clinical need [11]. Another urgent issue, still not completely clarified and a direct consequence of cisplatin-related survival increase, is represented by the incidence of second malignancies in GCTs survivors [12]. As consequence, national prevention and long-term follow-up policies for survivors have been promptly promoted worldwide [13].
2. Platinum Resistance and Sensitivity in Germ Cell Tumours
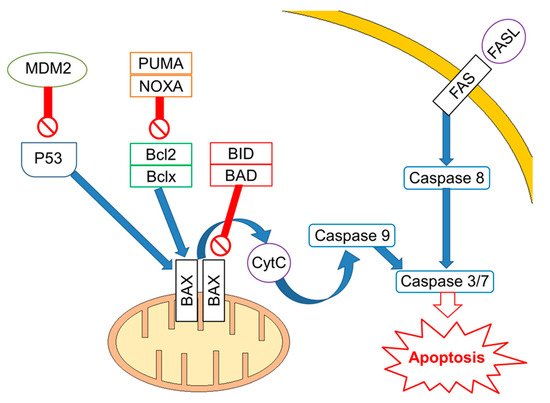
Mutations/losses of the tumour-suppressor TP53 and amplification/gains of its regulator, MDM2, as well as WNT/CTNNB1 pathway aberrations, which are involved in developmental processes and stemness, have been deeply investigated in platinum-resistant and metastatic disease. As already acknowledged, one of the hallmarks of GCTs is the presence of wild-type (WT) tp53 in most patients, although mutations in tp53 have been described in a few cases in the past [14] and more recently in a small subset (~7–15%) of cisplatin-resistant or relapsed GCT patients [15][16]. Activity of TP53 is regulated by the E3 ubiquitin ligase MDM2, which inhibits transcription of target genes by TP53 within direct binding to the transactivation domain of TP53 and through targeting TP53 for proteasome degradation by ubiquitination [17][18]. In physiological conditions, MDM2, through an auto-regulatory negative feedback, keeps TP53 levels low. Genomic alterations affecting both TP53 and MDM2 have been described in resistant GCTs [19]. A large study assessed 180 GCTs using whole exome sequencing, identifying MDM2/TP53 alterations exclusively in cisplatin-resistant tumours [16]. Notably, mutually exclusive TP53/MDM2 alterations were significantly more frequent among patients with unfavourable clinical characteristics, according to the IGCCCG poor-risk group [20] and having a mediastinal non-seminoma primary tumour site. The majority of MDM2 amplifications were observed in post-treatment samples, suggesting that tumour cells are selected during treatment [16]. Since tp53 mutations are rare, MDM2 amplification is a possible selection mechanism to prevent cell cycle arrest and DNA repair during the progression of disease. This hypothesis is reinforced by functional studies within cisplatin-sensitive and resistant testis cancer cell lines, indicating that the interaction between TP53 and MDM2 needed higher doses of cisplatin to be disrupted in resistant ones [21]. As previously reported, although most GCTs express WT TP53, posttranscriptional modifications are able to suppress the pro-apoptotic activity of TP53. For example, lysine methylation at the carboxyl terminus of TP53 represses its transcriptional activity, as demonstrated by reduced expression of PUMA and p21 in TC cells [22]. Furthermore, the expression of miR-372 and miR-373 was reported to block TP53 signalling, and their elevated expression levels have been detected in cisplatin-resistant GCT cell lines [23][24][25]. Another posttranslational modification is represented by deacetylation of TP53 by SIRT1, which may potentially repress TP53 activity in TGCT, suggesting that SIRT1 acts as an oncogene. However, the exact role of SIRT1 impacting TP53 activity in the context of TGCT has not be yet completely clarified since it has been reported as upregulated in various malignancies and acting as a tumour suppressor in other reports [26]. Moreover, the extrinsic apoptosis pathway via the interaction between FAS and FAS ligand is also activated by cisplatin, which increases expression of the FAS death receptor, a transcriptional target of TP53 [21][27]. Other studies presented the use of cisplatin results in an increased expression of pro-apoptotic proteins PUMA and NOXA, which are involved in the intrinsic apoptosis pathway [28][29][30]. Additionally, high protein levels of the pluripotency factor OCT4, typical for the embryonal carcinoma histotype, are associated with a high expression of NOXA, while OCT4 knockdown resulted in NOXA decrease [30]. Notably, high NOXA expression has been linked to TGCT, with good prognosis and embryonal carcinoma histology [31]. Crosstalk between the extrinsic and intrinsic apoptosis pathways, where FAS receptor activation results in caspase-8-mediated cleavage of BID, may further reinforce the apoptotic response. In a comprehensive genomic study, Taylor-Weiner et al. showed that primary TGCTs are uniformly wild type for TP53 and, by functional measurement of apoptotic signalling (BH3 profiling), they have demonstrated high mitochondrial priming that facilitates chemotherapy-induced apoptosis [32]. (Figure 1).
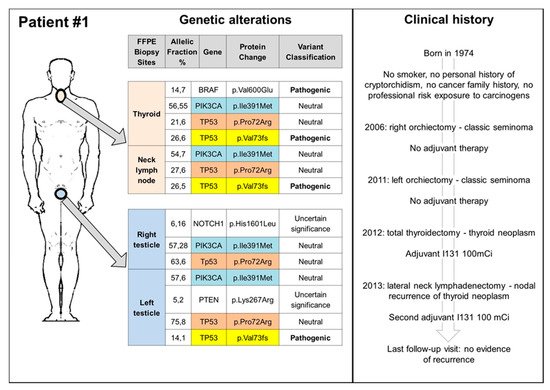
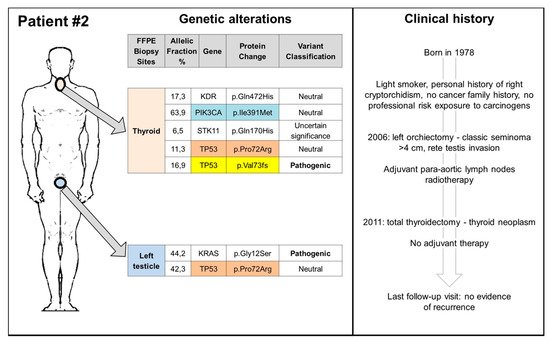
3. Germ Cell Tumours and Second Cancers
4. Conclusions
References
- Albers, P.; Albrecht, W.; Algaba, F.; Bokemeyer, C.; Cohn-Cedermark, G.; Fizazi, K.; Horwich, A.; Laguna, M.P.; Nicolai, N.; Oldenburg, J. Guidelines on Testicular Cancer: 2015 Update. Eur. Urol. 2015, 68, 1054–1068.
- Richardson, L.C.; Neri, A.J.; Tai, E.; Glenn, J.D. Testicular cancer: A narrative review of the role of socioeconomic position from risk to survivorship. Urol. Oncol. Semin. Orig. Investig. 2012, 30, 95–101.
- Ylönen, O.; Jyrkkiö, S.; Pukkala, E.; Syvänen, K.; Boström, P.J. Time trends and occupational variation in the incidence of testicular cancer in the Nordic countries. BJU Int. 2018, 122, 384–393.
- Condello, C.; Rescigno, P.; Ottaviano, M.; Nappi, L.; Tortora, M.; De Placido, S.; Palmieri, G. Clinical features and psychological aspects of the decision-making process in stage I testicular germ cell tumors. Futur. Oncol. 2018, 14, 1591–1599.
- Verhoeven, R.; Coebergh, J.; Kiemeney, L.; Koldewijn, E.; Houterman, S. Testicular cancer: Trends in mortality are well explained by changes in treatment and survival in the southern Netherlands since 1970. Eur. J. Cancer 2007, 43, 2553–2558.
- Richiardi, L.; Scélo, G.; Boffetta, P.; Hemminki, K.; Pukkala, E.; Olsen, J.H.; Weiderpass, E.; Tracey, E.; Brewster, D.; McBride, M.L.; et al. Second malignancies among survivors of germ-cell testicular cancer: A pooled analysis between 13 cancer registries. Int. J. Cancer 2007, 120, 623–631.
- Murty, V.V.; Chaganti, R.S. A genetic perspective of male germ cell tumors. Semin. Oncol. 1998, 25, 133–144.
- Adra, N.; Einhorn, L.H. Testicular cancer update. Clin. Adv. Hematol. Oncol. 2017, 15, 386–396.
- Weidner, N. Germ-cell tumors of the mediastinum. Semin. Diagn. Pathol. 1999, 16, 42–50.
- Bokemeyer, C.; Nichols, C.R.; Droz, J.-P.; Schmoll, H.-J.; Horwich, A.; Gerl, A.; Fossa, S.D.; Beyer, J.; Pont, J.; Kanz, L.; et al. Extragonadal Germ Cell Tumors of the Mediastinum and Retroperitoneum: Results from an International Analysis. J. Clin. Oncol. 2002, 20, 1864–1873.
- Varmus, H.; Harlow, E. Science funding: Provocative questions in cancer research. Nature 2012, 481, 436–437.
- Robinson, D.; Moller, H.; Horwich, A. Mortality and incidence of second cancers following treatment for testicular cancer. Br. J. Cancer 2007, 96, 529–533.
- Banna, G.L.; Nicolai, N.; Palmieri, G.; Ottaviano, M.; Balzarini, L.; Barone, D.; Basso, U.; Bavila, A.; Bertoni, F.; Calliada, F.; et al. ☆Corrigendum to “Recommendations for surveillance and follow-up of men with testicular germ cell tumors: A multidisciplinary consensus conference by the Italian Germ cell cancer Group and the Associazione Italiana di Oncologia Medica”. Crit. Rev. Oncol. Hematol. 2020, 146, 102865.
- Houldsworth, J.; Xiao, H.; Murty, V.; Chen, W.; Ray, B.; Reuter, V.E.; Bosl, G.J.; Chaganti, R. Human male germ cell tumor resistance to cisplatin is linked to TP53 gene mutation. Oncogene 1998, 16, 2345–2349.
- Loveday, C.; Litchfield, K.; Proszek, P.Z.; Cornish, A.J.; Santo, F.; Levy, M.; MacIntyre, G.; Holryod, A.; Broderick, P.; Dudakia, D.; et al. Genomic landscape of platinum resistant and sensitive testicular cancers. Nat. Commun. 2020, 11, 1–12.
- Bagrodia, A.; Lee, B.H.; Lee, W.; Cha, E.K.; Sfakianos, J.P.; Iyer, G.; Pietzak, E.J.; Gao, S.P.; Zabor, E.C.; Ostrovnaya, I.; et al. Genetic Determinants of Cisplatin Resistance in Patients with Advanced Germ Cell Tumors. J. Clin. Oncol. 2016, 34, 4000–4007.
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604.
- Romano, F.J.; Rossetti, S.; Conteduca, V.; Schepisi, G.; Cavaliere, C.; Di Franco, R.; La Mantia, E.; Castaldo, L.; Nocerino, F.; Ametrano, G.; et al. Role of DNA repair machinery and p53 in the testicular germ cell cancer: A review. Oncotarget 2016, 7, 85641–85649.
- Lafin, J.; Bagrodia, A.; Woldu, S.; Amatruda, J.F. New insights into germ cell tumor genomics. Andrology 2019, 7, 507–515.
- International Germ Cell Cancer Collaborative Group. International Germ Cell Consensus Classification: A prognostic fac-tor-based staging system for metastatic germ cell cancers. J. Clin. Oncol. 1997, 15, 594–603.
- Koster, R.; Timmer-Bosscha, H.; Bischoff, R.; Gietema, J.; De Jong, S. Disruption of the MDM2–p53 interaction strongly potentiates p53-dependent apoptosis in cisplatin-resistant human testicular carcinoma cells via the Fas/FasL pathway. Cell Death Dis. 2011, 2, e148.
- Zhu, J.; Dou, Z.; Sammons, M.A.; Levine, A.J.; Berger, S.L. Lysine methylation represses p53 activity in teratocarcinoma cancer cells. Proc. Natl. Acad. Sci. USA 2016, 113, 9822–9827.
- Voorhoeve, P.M.; le Sage, C.; Schrier, M.; Gillis, A.J.; Stoop, H.; Nagel, R.; Liu, Y.-P.; van Duijse, J.; Drost, J.; Griekspoor, A.; et al. A Genetic Screen Implicates miRNA-372 and miRNA-373 As Oncogenes in Testicular Germ Cell Tumors. Cell 2006, 124, 1169–1181.
- Duale, N.; Lindeman, B.; Komada, M.; Olsen, A.-K.; Andreassen, A.; Soderlund, E.J.; Brunborg, G. Molecular portrait of cis-platin induced response in human testis cancer cell lines based on gene expression profiles. Mol. Cancer 2007, 6, 53.
- Port, M.; Glaesener, S.; Ruf, C.; Riecke, A.; Bokemeyer, C.; Meineke, V.; Honecker, F.; Abend, M. Micro-RNA expression in cisplatin resistant germ cell tumor cell lines. Mol. Cancer 2011, 10, 52.
- Yi, J.; Luo, J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2010, 1804, 1684–1689.
- Spierings, D.; De Vries, E.; Vellenga, E.; De Jong, S. Loss of drug-induced activation of the CD95 apoptotic pathway in a cispla-tin-resistant testicular germ cell. Cell Death Differ. 2003, 10, 808–822.
- Mueller, T.; Voigt, W.; Simon, H.; Fruehauf, A.; Bulankin, A.; Grothey, A.; Schmoll, H.-J. Failure of activation of caspase-9 induces a higher threshold for apoptosis and cisplatin resistance in testicular cancer. Cancer Res. 2003, 63, 513–521.
- Gutekunst, M.; Oren, M.; Weilbacher, A.; Dengler, M.A.; Markwardt, C.; Thomale, J.; Aulitzky, W.E.; van der Kuip, H. p53 hypersensitivity is the predominant mechanism of the unique responsiveness of testicular germ cell tumor (TGCT) cells to cis-platin. PLoS ONE 2011, 6, e19198.
- Gutekunst, M.; Mueller, T.; Weilbacher, A.; Dengler, M.A.; Bedke, J.; Kruck, S.; Oren, M.; Aulitzky, W.E.; Van Der Kuip, H. Cisplatin Hypersensitivity of Testicular Germ Cell Tumors Is Determined by High Constitutive Noxa Levels Mediated by Oct-4. Cancer Res. 2013, 73, 1460–1469.
- Grande, L.; Bretones, G.; Rosa-Garrido, M.; Martin, E.M.G.; Hernandez, T.; Fraile, S.; Botella, L.; de Alava, E.; Vidal, A.; del Muro, X.G.; et al. Transcription Factors Sp1 and p73 Control the Expression of the Proapoptotic Protein NOXA in the Response of Testicular Embryonal Carcinoma Cells to Cisplatin. J. Biol. Chem. 2012, 287, 26495–26505.
- Taylor-Weiner, A.; Zack, A.T.-W.T.; O’Donnell, E.; Guerriero, J.L.; Bernard, B.; Reddy, A.; Han, G.C.; AlDubayan, S.H.; Amin-Mansour, A.; Schumacher, S.E.; et al. Genomic evolution and chemoresistance in germ-cell tumours. Nat. Cell Biol. 2016, 540, 114–118.
- Hellesnes, R.; Kvammen, Ø.; Myklebust, T.Å.; Bremnes, R.M.; Karlsdottir, Á.; Negaard, H.F.S.; Tandstad, T.; Wilsgaard, T.; Fosså, S.D.; Haugnes, H.S. Continuing increased risk of second cancer in long-term testicular cancer survivors after treatment in the cisplatin era. Int. J. Cancer 2019, 147, 21–32.


