Parkinson’s disease (PD) is the most common neurodegenerative movement disorder without any objective biomarker available to date. Increasing evidence highlights the critical role of neuroinflammation, including T cell responses, and spreading of aggregated α-synuclein in PD progression. Lymphocyte-activation gene 3 (LAG3) belongs to the immunoglobulin (Ig) superfamily expressed by peripheral immune cells, microglia and neurons and plays a key role in T cell regulation. The role of LAG3 has been extensively investigated in several human cancers, whereas until recently, the role of LAG3 in the central nervous system (CNS) has been largely unknown. Accumulating evidence highlights the potential role of LAG3 in PD pathogenesis, mainly by binding to α-synuclein fibrils and affecting its endocytosis and intercellular transmission, which sheds more light on the connection between immune dysregulation and α-synuclein spreading pathology. Serum and cerebrospinal fluid (CSF) soluble LAG3 (sLAG3) levels have been demonstrated to be potentially associated with PD development and clinical phenotype, suggesting that sLAG3 could represent an emerging PD biomarker. Specific single nucleotide polymorphisms (SNPs) of the LAG3 gene have been also related to PD occurrence especially in the female population, enlightening the pathophysiological background of gender-related PD clinical differences. Given also the ongoing clinical trials investigating various LAG3-targeting strategies in human diseases, new opportunities are being developed for PD treatment research.
1. Introduction
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder affecting approximately 1–2% of the population above the age of 65 years
[1]. The key pathological hallmarks of the disease involve the intracellular accumulation of α-synuclein in the form of Lewy bodies and Lewy dendrites, as well as the dopaminergic neuronal loss in the substantia pars compacta (SNpc) resulting in nigrostriatal degeneration
[1][2][3]. However, its pathology progresses gradually from the brainstem to cortical regions
[4].
Although many genetic and environmental factors have been associated with PD development, the exact etiology of the disease remains obscure. Along with autophagy impairment, mitochondrial dysregulation and oxidative damage, accumulating evidence suggest that neuroinflammation, including T cell-driven immune responses, as well as neuron-to-neuron transmission of aggregated α-synuclein may significantly contribute to PD progression
[5][6]. Given the absence of a reliable, objective and easily measured biomarker for PD, a growing number of studies have been focusing on the investigation of novel diagnostic and prognostic biomarker candidates, such as α-synuclein, dopamine metabolites, neuropeptide hormones like orexin, apolipoprotein A1, DJ-1, various molecules associated with proteasomes and micro-RNAs, as well as proinflammatory cytokines
[1][7]. However, the results of many of these studies are inconsistent, and there is still no available serum or cerebrospinal fluid (CSF) molecule that could be effectively used for PD diagnosis or progression
[1].
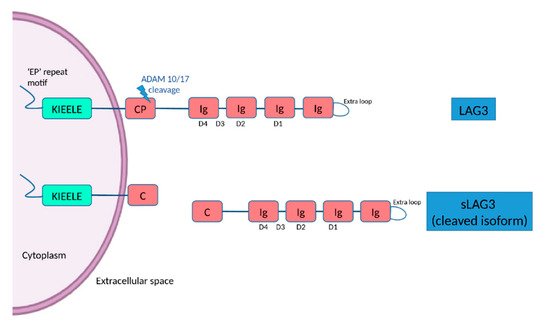
Lymphocyte-activation gene 3 (
LAG3, also termed as CD223) is located to chromosome 12p13 and belongs to the immunoglobulin (Ig) superfamily expressed by peripheral immune cells including subsets of natural killer cells, CD4
+ and CD8
+ T cells, B cells and dendritic cells, as well as microglial cells and neurons
[8][9]. LAG3, firstly identified in 1990
[10], is a transmembrane protein consisting of 498 amino acids, with four extracellular Ig-like domains (D1–D4) and an intracytoplasmic part
[11]. The D1 domain contains a unique extra loop and a small amino acid sequence that confers binding to major histocompatibility complex (MHC) class II molecules (MHC II). The KIEELE motif, a unique amino acid sequence in the intracellular domain is required for the LAG3-mediated negative inhibitory signal on T cells whereas an ‘EP’ repeat motif mediates anchorage (
Figure 1)
[11]. The transmembrane connecting peptide (CP) of LAG3 can be cleaved by the metalloproteinases ADAM10 and ADAM17 to produce its soluble form, detectable in serum. LAG3 is a CD4 homologue, and binds to MHC class II molecules on antigen-presenting cells with higher affinity than CD4
[11][12]. Being an immune checkpoint receptor, LAG3 regulated T cell immune responses and immune homeostasis, mainly by inhibiting T cell activation and proliferation
[9]. LAG3 is highly expressed in peripheral immune organs including the spleen and the thymus, and also in the central nervous system (CNS)
[13]. LAG3 could be expressed on neuronal cells
[9], but also on microglia
[14]. Although there is evidence on the role of LAG3 in other disorders, especially autoimmune diseases and cancer
[11], the function of LAG3 in the CNS is largely unknown
[15], and only recently, some experimental studies have begun to shed light on its role in neurodegenerative diseases.
Figure 1. The structure of LAG3 and its cleaved soluble form (sLAG3). LAG3 is a transmembrane protein consisting of 498 amino acids, with four extracellular Ig-like domains (D1–D4) and an intracellular domain, containing an ‘EP’ (glutamic acid-proline) repeat responsible for anchorage and a KIEELE motif, which is required for the LAG3-mediated negative inhibitory signal. LAG-3 can be cleaved within the transmembrane domain at the connecting peptide (CP) by two members of metalloproteases known as ADAM 10 and ADAM 17 to release sLAG-3, which contributes to the regulatory function of LAG-3.
2. The Implication of LAG3 in PD Pathogenesis
Increasing evidence highlights the fact that proteins traditionally considered to act in the peripheral immune system may actually play critical roles in the CNS and subsequently be implicated in neurological disorders
[15]. Being a mediator of T cell regulation and immune response, LAG3 has been recently suggested to be involved in PD pathogenesis and specifically in α-synuclein transmission.
Notably, a study that used preformed fibrils (PFFs) of recombinant mouse α-synuclein as a model to investigate the neuron-to-neuron transmission of α-synuclein, has demonstrated that LAG3 may play an important role in this process
[15]. Among the three identified transmembrane protein candidates for binding to exogenous α-synuclein PPFs after the unbiased screening, LAG3 exhibited the highest selectivity for α-synuclein PPFs compared to α-synuclein monomers
[15]. Further analysis revealed that out of the four Ig-like domains of LAG3, D1 was responsible for binding to α-synuclein PPFs, particularly via its residues 52–109
[15]. LAG3 also triggered α-synuclein PFFs endocytosis, since LAG3 deletion could significantly reduce the internalization of α-synuclein PFFs in mouse cortical neurons
[15]. The early endosomal marker RAB5, a small Ras-like GTPase, colocalized with α-synuclein PFFs in this experiment
[15], and there has been already reported that Rab5 is fundamentally implicated in α-synuclein endocytosis
[16]. Besides that, LAG3 overexpression enhanced the phosphorylation of α-synuclein at serine 129
[15], a process that has been already associated with PD pathology
[17]. Furthermore, α-synuclein PPFs-induced synaptic disruption was inhibited in LAG3 knockout in vitro, since the reduction of SNAP25 and synapsin II was prevented compared to wild-type cells
[15]. LAG3 deletion inhibited α-synuclein neuronal transmission, α-synuclein PPFs-induced neuronal loss and toxicity in vitro, accompanied by lower intracellular calcium levels
[15]. In particular, signalling via the intracellular domain of LAG3 could possibly mediate α-synuclein-induced neurotoxicity, since the deletion of this domain was found to reduce α-synuclein PPFs-induced toxic effects
[15]. LAG3 deletion or the usage of LAG3 antibodies also decreased the α-synuclein-PPFs-induced neurotoxicity in human A53T α-synuclein transgenic neuronal cell cultures, further highlighting the implication of LAG3 in the progression of human α-synuclein pathology
[15]. Additionally, LAG3 was expressed only in neurons and not in microglia or astrocytes in this study, suggesting that the abovementioned proposed mechanism for α-synuclein transmission may be neuron-specific
[15].
Apart from in vitro evidence, LAG3 has been significantly involved in α-synuclein transmission and related neurotoxicity in vivo
[15]. Phosphorylated α-synuclein staining was significantly reduced in the dopaminergic neurons of SNpc after injection of α-synuclein PFFs into the striatum of LAG3 knockout mice, compared to wild-type ones
[15]. The α-synuclein PPFs-induced dopaminergic neuronal loss, along with the reduction of dopamine and its metabolites (3MT, DOPAC and HVA), as well as the decrease of TH and DAT enzymes were delayed in the SNpc of the LAG3 knockout animals
[15]. Of note, the behavioral impairment of the animals was also prevented in the case of LAG3 deletion, as evaluated by grip strength and the pole test
[15] that is considered to effectively reflect dopaminergic function
[18]. Collectively, these interesting findings suggest that LAG3 could bind to extracellular α-synuclein fibrils, be involved in the initiation of α-synuclein endocytosis and transmission, as well as contribute to α-synuclein-induced dopaminergic neuronal loss and neurotoxicity. Therefore, LAG3 protein may play a fundamental role in α-synuclein spreading pathology and neurodegeneration in PD, and further studies are needed to investigate the exact underlying molecular mechanisms.
Parkinson’s disease shares some common features with prion diseases, including abnormal protein aggregation, neuronal loss and microglial activation. However, a recent in vivo study has shown that although LAG3 levels were increased after prion infection in mice, LAG3 knockout did not affect PrPSc load, microglia activation, astrocyte reaction, expression of inflammatory genes and degree of neurodegeneration in the animal models, suggesting that LAG3 may not be implicated in the pathogenesis of prion diseases
[9]. Nevertheless, α-synuclein and PrPSc differ significantly in terms of molecular structure and function, and the underlying pathophysiology of PD and prion diseases, including Creutzfeldt–Jakob disease (CJD) is not identical
[19]. The lack of LAG3 involvement in the pathogenesis of prion disease may further support the fact that it is specific for PD, and potentially able to differentiate from “pure” prionopathies, such as CJD.
3. LAG3 as a Potential Biomarker for PD
The sLAG3 is produced either by alternative splicing of LAG3 RNA or cleaved and released from the full-length LAG3 protein
[12][20]. A recent study in the Chinese population has indicated that serum sLAG3 levels measured by quantitative enzyme-linked immunosorbent assay (ELISA) were significantly higher in PD patients compared to age- and gender-matched controls and to patients with essential tremor, another common movement disorder
[7]. Its estimated sensitivity and specificity was about 82% and 78% versus controls, and 76% and 82% versus patients with essential tremor, respectively
[7]. Increased serum sLAG3 levels were also found to be associated with more severe non-motor symptoms as evaluated by non-motor symptoms scale (NMSS) scores, as well as excessive daytime sleepiness
[7]. In this regard, increased CSF levels of YKL-40, an inflammatory marker, have been correlated with faster cognitive impairment in PD
[21], and the concentration of tumor necrosis factor-α, interleukin-1β and nitric oxide in the CSF have been associated with probable REM sleep behavior disorder in PD patients
[22]. These findings suggest that excessive inflammation may accompany non-motor symptoms of PD, and LAG3, as an indicator of cellular immune responses, may reflect the increased inflammation related to non-motor PD manifestations
[7]. Taken together, apart from its value as a diagnostic biomarker, serum sLAG3 may be also associated with PD clinical phenotypes.
Based on these interesting pieces of evidence, another larger study aimed to additionally explore the potential role of sLAG3 in the CSF as a potential PD biomarker in a Chinese cohort
[23]. More specifically, it was demonstrated that the mean sLAG3 levels in CSF were lower in PD cases compared to healthy controls, whereas no significant differences were observed in the case of serum sLAG3 levels between these groups
[23]. These results are in disagreement with the findings of the previously mentioned study, in which serum sLAG3 levels were increased in PD patients compared to controls. This discrepancy could be at least partially explained by the different methods used for the evaluation of sLAG3 levels in each study (ELISA versus mesoscale discovery electrochemiluminescence immunoassay (MSD-ECL). Indeed, the MSD-ECL technology displays higher repeatability, stability, sensitivity and homogeneity than ELISA assay
[24]. Gender, age, disease duration and severity as assessed by Hoehn and Yahr PD staging, did not significantly affect sLAG3 concentration in CSF of PD patients
[23]. The estimated sensitivity and specificity of CSF sLAG3 as a possible PD biomarker were about 45% and 90%, respectively
[23]. Although the sensitivity is low, this is the result of a single study applying MSD-ECL in CSF samples, where the concentration of the antigen may be low or the affinity reagents may need better standardization. However, the high estimated specificity of CSF sLAG3 is very encouraging
[23].
Although the results of studies investigating the role of α-synuclein as a potential biomarker are inconsistent, several lines of evidence have shown that CSF α-synuclein levels may be lower in PD patients compared to controls
[25][26]. Given the possible implication of LAG3 in α-synuclein propagation, the relationship between sLAG3 and α-synuclein levels in the CSF has been also investigated
[23]. In particular, although CSF sLAG3 levels were positively correlated with the CSF levels of α-synuclein in controls, no significant associations were found in PD patients
[23]. It is important to mention that the two subtypes of LAG3 protein (full-length LAG3 and sLAG3) may exert distinct roles in immune regulation. More specifically, full-length membrane LAG3 generally suppresses T cell immune responses
[27], whereas membrane LAG3 blockade or proteolytic cleavage, induces T cell activation and production of cytokines
[28]. sLAG3 has been also demonstrated to inhibit the differentiation of monocytes to both macrophages and dendritic cells
[29], whereas LAG3 blockade has been reported to act as an enhancer of T cell response in the case of antitumor cell vaccination
[30]. Although the exact roles of LAG3 and sLAG3 in the CNS and PD are still unknown, based on the results of the study by Guo and colleagues, it has been speculated that LAG3 and sLAG3 imbalance in the brain may create an immunologically impaired microenvironment, susceptible to α-synuclein-mediated neurodegeneration, which the aggregated α-synuclein could utilize in order to spread in the CNS
[23].
The occipital lobes were reported to exhibit the greatest atrophy in this study, followed by the temporal lobes and basal ganglia
[7]. Interestingly, visual hallucinations were found to be related to selective brain atrophy of the LAG3 high expressing cuneus and lingual gyrus of the occipital lobe in the examined PD population, and these brain regions have already been associated with PD-related hallucinations in previous reports
[31]. The degree of cognitive decline in PD has been associated with the degree of temporal atrophy in other studies
[32]. Collectively, these combined imaging-genetic findings suggest that the higher regional
LAG3 gene expression may be associated with regional atrophy in PD, and could also be correlated with the degree of non-motor manifestations, including psychotic symptoms, which are in agreement with the results of the abovementioned study investigating sLAG3 levels in the serum of PD patients
[7]. Notably, as imaging and genetic data were obtained from different populations (PD patients and healthy controls respectively) and the possibility that gene expression patterns may significantly differ in the case of PD, further studies are obviously needed to validate the results of the abovementioned study (
Figure 2).
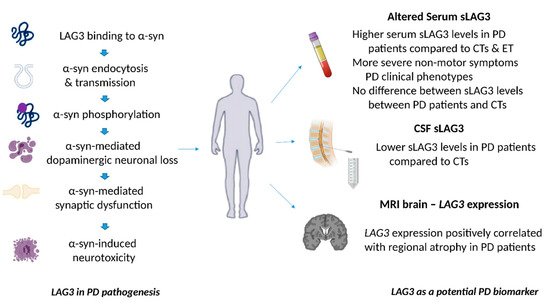
Figure 2. The role of LAG3 in the pathogenesis of Parkinson’s disease and its biomarker potential. LAG3 can bind to α-synuclein and trigger its endocytosis, contributing to α-synuclein cell-to-cell transmission. Overexpression of LAG3 may enhance the phosphorylation of α-synuclein at serine 129. LAG3 has been also implicated in α-synuclein dopaminergic neuronal loss, neurotoxicity and synaptic dysfunction. Clinical evidence has shown that soluble LAG3 (sLAG3) levels may be higher in patients with PD compared to controls (CTs) or patients with essential tremor (ET), whereas there is also evidence indicating that serum sLAG3 levels display no difference between PD patients and CTs. Lower sLAG3 levels in the cerebrospinal fluid (CSF) have been associated with PD development, and LAG3 gene expression has been also correlated with regional atrophy in magnetic resonance imaging (MRI) brain scans of patients with PD.
 +1 credit
+1 credit