 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Massimo Pancione | + 3166 word(s) | 3166 | 2021-06-25 08:44:09 | | | |
| 2 | Lindsay Dong | Meta information modification | 3166 | 2021-07-23 05:21:13 | | |
Video Upload Options
The centrosome plays a major role in organizing the microtubule cytoskeleton in animal cells regulating cellular architecture and cell division. Loss of centrosome integrity activates the p38-p53-p21 pathway, which results in cell-cycle arrest or senescence and acts as a cell-cycle checkpoint pathway. Structural and numerical centrosome abnormalities can lead to aneuploidy and chromosomal instability (CIN). New findings derived from studies on cancer and rare genetic disorders suggest that centrosome dysfunction alters the cellular microenvironment through Rho GTPases, p38, and JNK (c-Jun N-terminal Kinase)-dependent signaling in a way that is favorable for pro-invasive secretory phenotypes and aneuploidy tolerance.
1. Introduction
The overwhelming majority of cancer mortality is caused by metastasis, a complex process that remains the least understood aspect of cancer biology [1]. Metastasis is a process in which cancer cells disseminate from the primary tumor and seed new colonies at distant sites. It involves the local invasion of primary-tumor cells into surrounding tissue, intravasation of these cells into the circulatory system, and subsequent extravasation to other tissues through the vascular walls [1].
In this way, cancer cells travel to the parenchyma of a distant tissue and seed microscopic colonies that proliferate to form metastatic lesions. In addition to cancer cell-autonomous mechanisms, metastatic growth depends on the interactions of cancer cells with their niche microenvironment and the crosstalk with various stromal cells, including endothelial cells, fibroblasts, and cells from the innate and adaptive immune system [2]. Over the last years, the biological programs that underlie the dissemination and metastatic outgrowth of cancer cells have begun to emerge. An important aspect is the diversification and adaptation of cancer cells that can be achieved by two main biological processes (1) the dormancy programs (DP) characterized by the activation of quiescence and survival pathways and (2) epithelial-mesenchymal transition (EMT) in which epithelial cells lose their cell polarity and cell-cell adhesions, and gain migratory and invasive properties to become under certain conditions mesenchymal cells that sometimes present stem cell-like properties [2][3]. Notably, in addition to phenotypic differences, significant genotypic diversity exists within tumors, a process known as intra-tumor heterogeneity (ITH) that can be observed at the genetic, proteomic, morphological, and environmental level [4].
One of the central drivers of intra-tumor diversification is the chromosome copy number, instability of particular loci, large chromosome segments, or entire chromosomes. This instability may alter the chromosomal content of a cell (aneuploidy). Karyotypic heterogeneity in tumor cells derives from “chromosomal instability” (CIN), a hallmark that not only generates abnormal aneuploid karyotypes, but also expands continually phenotypic heterogeneity as tumor cell populations undergo consecutive cell divisions [5].
2. Aneuploidy and CIN: Two Sides to the Debate in Cancer
The link between chromosomal abnormalities and cancer was first proposed by the German biologist, Theodor Boveri, over a hundred years ago [6]. Decades of studies have shown that errors in mechanisms of cell division are an important source of genomic diversification that promotes ITH and cancer evolution. Paradoxically, despite the observation that aneuploidy is an unfavorable state for cancer cells, most healthy cells such as osteoclasts or hepatocytes are highly aneuploid [6]. Therefore, aneuploidy is increasingly recognized as a factor that might promote genetic diversity. Aneuploidy is present in around 80% of human solid neoplasms [7].
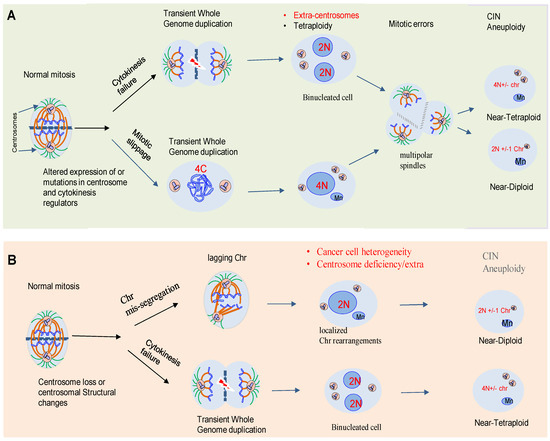
3. Centrosome Biology and CIN
In animal cells, the centrosome is the major microtubule-organizing center (MTOC) [6]. The core of the centrosome consists of centrioles of nine microtubule (MT) triplets embedded in an ordered and protein-rich matrix called pericentriolar material (PCM). Centrosomes promote the production of bipolar mitotic spindles and supply a matrix of primary cilia in various cell types (Figure 2A) [10][11]. In addition to these structural functions, centrosomes and primary cilia have also evolved into essential signaling hubs to build and regulate the sensory/motor characters of metazoans [12][13]. It is increasingly recognized that centrosome is an organizing unit not only for MTs, but also for actin, Golgi apparatus, and signaling molecules coordinating cell migration, cell polarity, and fundamental signal transduction pathways [14][15]. Large-scale proteomic studies have revealed that there are more than 400 centrosome-associated proteins (about 3% of human proteome, but the exact functions of most of these proteins are still unknown and require further investigation. However, recent advances in imaging, proteomics, and structural biology have revealed new insights into how microtubule-associated proteins regulate the structure, length, and stability of centrosome/cilium complex opening up new avenues for future research [6][15][16][17].
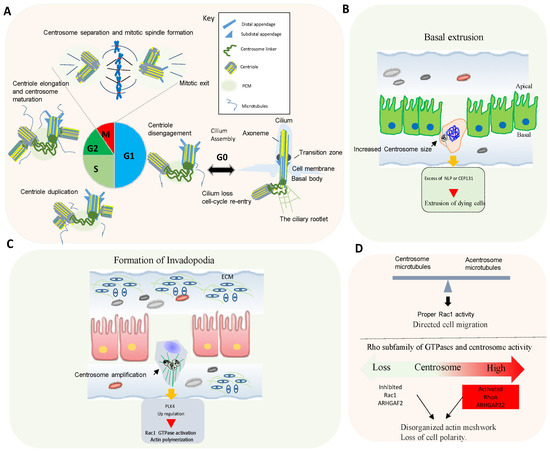
4. Rho GTPases Signaling and Centrosome Aberrations
Proteins controlling microtubule dynamics and processes that require changes in cell shape and motility are important for tumor dissemination. Rho GTPase signaling is commonly altered in human tumors, and an elevated expression and/or activation of Rho GTPases often correlates with tumor progression, metastasis, and poor prognosis [18].
Centrosome disruption induces excessive Rac1 activation around the cell periphery, causing rapid focal adhesion turnover, a disorganized actin network, randomly protruding lamellipodia, and the loss of cell polarity [19]. This supports that the centrosome integrity guides the spatial activation of Rac1 to control normal cell polarization and directed cell migration (Figure 2D).
Conversely, centrosome amplification in cultured cells also activates Rac1. In fact, in cells with extra centrosomes, increased centrosomal microtubule nucleation leads to Rac1 activation, disruption of cell-cell contacts, and invasive behavior [6][18].
5. Centrosome, Cell Cycle and Inflammatory Responses
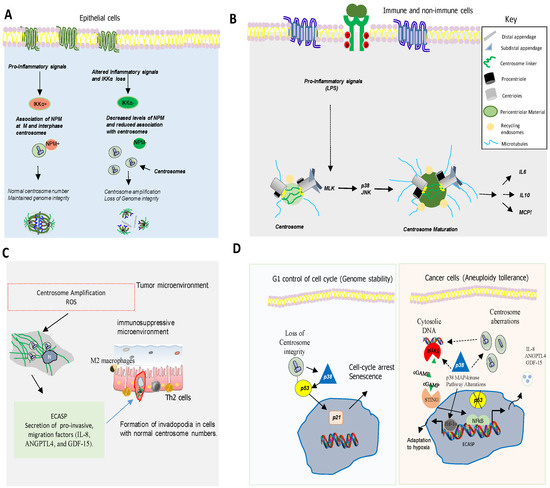
6. p38MAPK as a Key Mediator of Chromosome Stability and Cell Cycle
One of the main causes of CIN is the deregulation of the cell cycle [29]. p38MAPK regulates cell cycle in different situations, controlling genomic instability. For example, p38MAPK controls cell cycle at G0, G1/S, and G2/M transitions to ensure genetic integrity and stability of the cell at each step [29]. p38MAPK also regulates actin cytoskeleton organization, which is required for cytokinesis and mitosis [30][31]. Hence, p38α MAPK deficiency in hepatocytes induces actin disassembly and cytokinesis failure, which leads to the generation of genetically unstable polyploid cells [31]. Furthermore, inhibition of p38MAPK in combination with taxanes increases genomic instability and DNA damage, impairing DNA replication in breast cancer cells [32].
Several observations indicate that p38MAPK, a kinase activated by various types of stress [33][34] also plays a pivotal role in chromosome mis-segregation and aneuploidy tolerance upstream of p53 [35][36][37] (Figure 3D). This is supported by studies showing that pharmacological chemical inhibition of p38MAPK overcomes p53-dependent cell-cycle arrest after prolonged mitosis or chromosome mis-segregation [38][39] and enhances CIN [40]. It is known that activation of p38MAPK in response to DNA damage induces a G2/M cell cycle checkpoint to repair DNA through p53-dependent mechanisms [38][39][40] (Figure 3D).
Centrosome abnormalities are additional causes of CIN and p38MAPK has emerged as a kinase with a key role in centrosome dysfunction. Hence, the inhibition of p38MAPK rescued cell cycle progression after depletion of centrosome proteins [41]. Evidence also shows that p38MAPK activity is essential for centrosome normal functioning. For example, p38MAPK localizes at the mitotic centrosome allowing chromosomal segregation [41]. Furthermore, localization of p38α MAPK at the kinetochores and the centrosome is also essential for proper chromosomal segregation [42][41]. Active p38MAPK has also been localized at other structures such as the centriolar satellites, discovering a p38MAPK/MK2/14-3-3 signaling cascade that targets centrosome functions and modulates its response to cell stress [41]. All this evidence point to p38MAPK as a key component of a feedback pathway quality control that operates in mitosis and cell division to detect and transform chromosome alterations into a robust G1 arrest, often dependent on p53.
7. p38MAPKs in Aneuploidy, Inflammation and Immune Evasion
it is important to underline the relevant role of p38 MAPKs regulating the expression of pro-inflammatory molecules. p38α MAPK is involved in the induction of the expression of several inflammatory cytokines [43] such as TNF-α, IL-1, and IL-6 and other mediators of inflammation such as cyclooxygenase 2 (COX2), contributing to the development and progression of gliomas, breast cancer, head and neck squamous cell carcinoma, skin cancer or colorectal cancer (CRC) [44][45][46][47]. The regulation of these pro-inflammatory molecules by p38α pathway includes transcriptional and posttranscriptional mechanisms [47][48][49][50]. MK2, a p38α downstream kinase, plays a key role in this posttranscriptional regulation by stabilizing mRNAs and promoting translation [46]. For example, MK2 increases interleukin IL-6 expression through stabilization of its mRNA, while TNF-α production is enhanced by promoting its translation [48]. RNA binding proteins (RBDs), such as AUF-1, HuR (Human antigen R), and TTP (tristetraprolin) interact with mRNAs AU-rich sequences in the 3’Unstranslated region (UTR) to regulate their stability and MK2 controls the activity of these proteins [48]. In particular, TTP promotes mRNAs degradation, action inhibited by p38 and/or MK2 [51][52][53], for example, to increase TNF-α mRNA stability [54][55] and other mRNAs involved in inflammation and cancer growth such as COX-2 (Cyclooxygenase-2), VEGF (Vascular endothelial growth factor), and IL-10 [2].
8. Conclusions and Future Perspective
The existing evidence suggests that centrosomes play a key role in the regulation of cell senescence, an irreversible type of growth arrest that takes place when cells suffer extensive intrinsic and/or extrinsic damage. Centrosome dysfunction is inseparably linked to aneuploidy and CIN, both hallmarks of tumor cells. Recent advances indicate that centrosome defects (numerical and structural) could contribute to accelerate cancer cell immune evasion through different mechanisms. The inflammatory microenvironment could also result in aneuploidy and spread CIN in tumor cells by inducing a direct genotoxic stress and/or an epithelial–mesenchymal transition (EMT) process. This would lead to a feed-forward loop. Interestingly, the idea that the centrosome can also act as a key coordinator of cellular processes unrelated to microtubule organization, acting for example as a stress sensor, has emerged in recent years. Hence, in addition to well-known cellular stresses such as those induced by DNA damage, oxidative stress, centrosomes abnormalities can also regulate cell cycle arrest and cell senescence through the release of inflammatory mediators. Eukaryotic cell division is a central process that requires complex changes in cytoskeletal organization and function. In recent years, the relevance of Rho GTPases and p38 MAPK in the regulation of many aspects of cell cycle transition, mitosis, and cytokinesis is emerging. Many of these factors and processes have been associated with CIN and inflammation by activating the cGAS-STING pathway. However, the precise nature of these interactions to promote metastasis needs to be fully characterized. Emerging evidence associates CIN to both, promotion and suppression of anti-tumor immunity depending on the type and origin of the tumor. In addition to the well-characterized role of p38 in inflammation and immunity, cell cycle arrest is also regulated by p38, which directly or indirectly influences motor proteins, microtubule dynamics, and centrosome activity. Notably, increasing numbers of evidence points to p53 as a common protein among the different involved pathways. Therefore, in the next future, it will be of great interest to establish the functional significance of centrosomal p53 and p38 activity in relation to various structural centrosome proteins and kinases that regulate them under physiological and pathological conditions.
References
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 1–17.
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691.
- Zhuang, X.; Zhang, H.; Hu, G. Cancer and Microenvironment Plasticity: Double-Edged Swords in Metastasis. Trends Pharmacol. Sci. 2019, 40, 419–429.
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628.
- Da Silva-Diz, V.; Lorenzo-Sanz, L.; Bernat-Peguera, A.; Lopez-Cerda, M.; Muñoz, P. Cancer cell plasticity: Impact on tumor pro-gression and therapy response. Semin Cancer Biol. 2018, 53, 48–58.
- Remo, A.; Li, X.; Schiebel, E.; Pancione, M. The Centrosome Linker and Its Role in Cancer and Genetic Disorders. Trends Mol. Med. 2020, 26, 380–393.
- Bakhoum, S.F.; Cantley, L.C. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018, 174, 1347–1360.
- Fox, D.T.; Soltis, D.E.; Soltis, P.S.; Ashman, T.-L.; Van de Peer, Y. Polyploidy: A Biological Force from Cells to Ecosystems. Trends Cell Biol. 2020, 30, 688–694.
- Marcozzi, A.; Pellestor, F.; Kloosterman, W.P. The Genomic Characteristics and Origin of Chromothripsis. Methods Mol. Biol. 2018, 1769, 3–19.
- Joukov, V.; De Nicolo, A. The Centrosome and the Primary Cilium: The Yin and Yang of a Hybrid Organelle. Cells 2019, 8, 701.
- Frye, K.; Renda, F.; Fomicheva, M.; Zhu, X.; Gong, L.; Khodjakov, A.; Kaverina, I. Cell Cycle-Dependent Dynamics of the Golgi-Centrosome Association in Motile Cells. Cells 2020, 9, 1069.
- Arslanhan, M.D.; Gulensoy, D.; Firat-Karalar, E.N. A Proximity Mapping Journey into the Biology of the Mammalian Cen-trosome/Cilium Complex. Cells 2020, 9, 1390.
- Uzbekov, R.; Alieva, I. Who are you, subdistal appendages of centriole? Open Biol. 2018, 8.
- Inoue, D.; Obino, D.; Pineau, J.; Farina, F.; Gaillard, J.; Guerin, C.; Blanchoin, L.; Lennon-Duménil, A.; Théry, M. Actin filaments regulate microtubule growth at the centrosome. EMBO J. 2019, 38.
- Uzbekov, R.E.; Avidor-Reiss, T. Principal Postulates of Centrosomal Biology. Version 2020. Cells 2020, 9, 2156.
- Conkar, D.; Firat-Karalar, E.N. Microtubule-associated proteins and emerging links to primary cilium structure, assembly, maintenance, and disassembly. FEBS J. 2021, 288, 786–798.
- Nigg, E.A.; Holland, A.J. Once and only once: Mechanisms of centriole duplication and their deregulation in dis-ease. Nat. Rev. Mol. Cell Biol. 2018, 19, 297–312.
- Svensmark, J.H.; Brakebusch, C. Rho GTPases in cancer: Friend or foe? Oncogene 2019, 38, 7447–7456.
- Kloc, M.; Uosef, A.; Wosik, J.; Kubiak, J.Z.; Ghobrial, R.M. RhoA Pathway and Actin Regulation of the Golgi/Centriole Complex. Results Probl Cell Differ. 2019, 67, 81–93.
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS–STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39.
- Liccardi, G.; Ramos Garcia, L.; Tenev, T.; Annibaldi, A.; Legrand, A.J.; Robertson, D.; Feltham, R.; Anderton, H.; Darding, M.; Peltzer, N.; et al. RIPK1 and Caspase-8 Ensure Chromosome Stability Inde-pendently of Their Role in Cell Death and Inflammation. Mol. Cell. 2019, 73, 413–428.
- Xia, X.; Liu, S.; Xiao, Z.; Zhu, F.; Song, N.-Y.; Zhou, M.; Liu, B.; Shen, J.; Nagashima, K.; Veenstra, T.D.; et al. An IKKα-Nucleophosmin Axis Utilizes Inflammatory Signaling to Promote Genome Integrity. Cell Rep. 2013, 5, 1243–1255.
- Vertii, A.; Ivshina, M.; Zimmerman, W.; Hehnly, H.; Kant, S.; Doxsey, S. The Centrosome Undergoes Plk1-Independent Interphase Maturation during Inflammation and Mediates Cytokine Release. Dev. Cell 2016, 37, 377–386.
- Lomastro, G.M.; Holland, A.J. The Emerging Link between Centrosome Aberrations and Metastasis. Dev. Cell 2019, 49, 325–331.
- Zulato, E.; Favaretto, F.; Veronese, C.; Campanaro, S.; Marshall, J.D.; Romano, S.; Cabrelle, A.; Collin, G.B.; Zavan, B.; Belloni, A.S.; et al. ALMS1-Deficient Fibroblasts Over-Express Extra-Cellular Matrix Components, Display Cell Cycle Delay and Are Resistant to Apoptosis. PLoS ONE 2011, 6, e19081.
- Huang, X.; Xiang, L.; Fang, X.; Liu, W.; Zhuang, Y.; Chen, Z.; Shen, R.; Cheng, W.; Han, R.; Zheng, S.; et al. Functional characterization of CEP250 variant identified in nonsyndromic retinitis pigmentosa. Hum. Mutat. 2019, 40, 1039–1045.
- Wu, W.; Zhou, H.; He, F.; Xiao, Z.; Jiang, Y.; Zhao, M. Arsenate-mediated G2 cell cycle arrest in U-2OS cells involves phosphory-lation of human polycomb protein 2 by p38 MAPK. FEBS Lett. 2018, 592, 4087–4097.
- Mikule, K.; Delaval, B.; Kaldis, P.; Jurcyzk, A.; Hergert, P.; Doxsey, S. Loss of centrosome integrity induces p38-p53-p21-dependent G1-S arrest. Nat. Cell Biol. 2007, 9, 160–170.
- Jallepalli, P.V.; Lengauer, C. Chromosome segregation and cancer: Cutting through the mystery. Nat. Rev. Cancer 2001, 1, 109–117.
- Cuenda, A.; Rousseau, S. p38 MAP-Kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta (BBA) Bioenerg. 2007, 1773, 1358–1375.
- Tormos, A.M.; Rius-Perez, S.; Jorques, M.; Rada, P.; Ramirez, L.; Valverde Ángela, M.; Nebreda Ángel, R.; Sastre, J.; Taléns-Visconti, R. p38α regulates actin cytoskeleton and cytokinesis in hepatocytes during development and aging. PLoS ONE 2017, 12, e0171738.
- Cánovas, B.; Igea, A.; Sartori, A.A.; Gomis, R.R.; Paull, T.T.; Isoda, M.; Pérez-Montoyo, H.; Serra, V.; González-Suárez, E.; Stracker, T.H.; et al. Targeting p38α Increases DNA Damage, Chromosome Instability, and the Anti-tumoral Response to Taxanes in Breast Cancer Cells. Cancer Cell 2018, 33, 1094–1110.e8.
- Nebreda, A.R.; Porras, A. p38 MAP kinases: Beyond the stress response. Trends Biochem. Sci. 2000, 25, 257–260.
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417.
- Takenaka, K.; Moriguchi, T.; Nishida, E. Activation of the protein kinase p38 in the spindle assembly checkpoint and mitotic arrest. Science 1998, 280, 599–602.
- Tang, J.; Yang, X.; Liu, X. Phosphorylation of Plk1 at Ser326 regulates its functions during mitotic progression. Oncogene 2008, 27, 6635–6645.
- Lee, K.; Kenny, A.E.; Rieder, C.L. P38 Mitogen-activated Protein Kinase Activity Is Required during Mitosis for Timely Satisfaction of the Mitotic Checkpoint But Not for the Fidelity of Chromosome Segregation. Mol. Biol. Cell 2010, 21, 2150–2160.
- Thompson, S.L.; Compton, D.A. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol. 2010, 188, 369–381.
- Uetake, Y.; Sluder, G. Cell-cycle progression without an intact microtuble cytoskeleton. Curr. Biol. 2007, 17, 2081–2086.
- Bulavin, D.V.; Higashimoto, Y.; Popoff, I.J.; Gaarde, W.A.; Basrur, V.; Potapova, O.; Appella, E.; Fornace, A.J., Jr. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nat. Cell Biol. 2001, 411, 102–107.
- Tollenaere, M.A.X.; Villumsen, B.H.; Blasius, M.; Nielsen, J.C.; Wagner, S.A.; Bartek, J.; Beli, P.; Mailand, N.; Bekker-Jensen, S. p38- and MK2-dependent signalling promotes stress-induced centriolar satellite remodelling via 14-3-3-dependent sequestration of CEP131/AZI1. Nat. Commun. 2015, 6, 10075.
- Kukkonen-Macchi, A.; Sicora, O.; Kaczynska, K.; Oetken-Lindholm, C.; Pouwels, J.; Laine, L.; Kallio, M.J. Loss of p38gamma MAPK induces pleiotropic mitotic defects and massive cell death. J. Cell Sci. 2011, 124, 216–227.
- Wagner, E.F.; Nebreda Ángel, R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549.
- Timoshenko, A.V.; Chakraborty, C.; Wagner, G.F.; Lala, P.K. COX-2-mediated stimulation of the lymphangiogenic factor VEGF-C in human breast cancer. Br. J. Cancer 2006, 94, 1154–1163.
- Xu, K.; Shu, H.-K.G. EGFR Activation Results in Enhanced Cyclooxygenase-2 Expression through p38 Mitogen-Activated Protein Kinase–Dependent Activation of the Sp1/Sp3 Transcription Factors in Human Gliomas. Cancer Res. 2007, 67, 6121–6129.
- Ray, A.L.; Berggren, K.L.; Cruz, S.R.; Gan, G.N.; Beswick, E.J. Inhibition of MK2 suppresses IL-1β, IL-6, and TNF-α-dependent colorectal cancer growth. Int. J. Cancer 2018, 142, 1702–1711.
- Soni, S.; Saroch, M.K.; Chander, B.; Tirpude, N.V.; Padwad, Y.S. MAPKAPK2 plays a crucial role in the progression of head and neck squamous cell carcinoma by regulating transcript stability. J. Exp. Clin. Cancer Res. 2019, 38, 1–13.
- Neininger, A.; Kontoyiannis, D.; Kotlyarov, A.; Winzen, R.; Eckert, R.; Volk, H.-D.; Holtmann, H.; Kollias, G.; Gaestel, M. MK2 Targets AU-rich Elements and Regulates Biosynthesis of Tumor Necrosis Factor and Interleukin-6 Independently at Different Post-transcriptional Levels. J. Biol. Chem. 2002, 277, 3065–3068.
- Brook, M.; Tchen, C.R.; SantaLucia, T.; McIlrath, J.; Arthur, J.S.C.; Saklatvala, J.; Clark, A.R. Posttranslational Regulation of Tristetraprolin Subcellular Localization and Protein Stability by p38 Mitogen-Activated Protein Kinase and Extracellular Signal-Regulated Kinase Pathways. Mol. Cell. Biol. 2006, 26, 2408–2418.
- Marchese, F.P.; Aubareda, A.; Tudor, C.; Saklatvala, J.; Clark, A.R.; Dean, J.L.E. MAPKAP Kinase 2 Blocks Tristetraprolin-directed mRNA Decay by Inhibiting CAF1 Deadenylase Recruitment. J. Biol. Chem. 2010, 285, 27590–27600.
- Clement, S.L.; Scheckel, C.; Stoecklin, G.; Lykke-Andersen, J. Phosphorylation of Tristetraprolin by MK2 Impairs AU-Rich Element mRNA Decay by Preventing Deadenylase Recruitment. Mol. Cell. Biol. 2010, 31, 256–266.
- Mahtani, K.R.; Brook, M.; Dean, J.L.E.; Sully, G.; Saklatvala, J.; Clark, A.R. Mitogen-Activated Protein Kinase p38 Controls the Expression and Posttranslational Modification of Tristetraprolin, a Regulator of Tumor Necrosis Factor Alpha mRNA Stability. Mol. Cell. Biol. 2001, 21, 6461–6469.
- Hitti, E.; Iakovleva, T.; Brook, M.; Deppenmeier, S.; Gruber, A.D.; Radzioch, D.; Clark, A.R.; Blackshear, P.J.; Kotlyarov, A.; Gaestel, M. Mitogen-Activated Protein Kinase-Activated Protein Kinase 2 Regulates Tumor Necrosis Factor mRNA Stability and Translation Mainly by Altering Tristetraprolin Expression, Stability, and Binding to Adenine/Uridine-Rich Element. Mol. Cell. Biol. 2006, 26, 2399–2407.
- Lai, W.S.; Parker, J.S.; Grissom, S.F.; Stumpo, D.J.; Blackshear, P.J. Novel mRNA Targets for Tristetraprolin (TTP) Identified by Global Analysis of Stabilized Transcripts in TTP-Deficient Fibroblasts. Mol. Cell. Biol. 2006, 26, 9196–9208.
- Del Reino, P.; Alsina-Beauchamp, D.; Escós, A.; Cerezo-Guisado, M.I.; Risco, A.; Aparicio, N.; Zur, R.; Fernandez-Estévez, M.; Collantes, E.; Montans, J.; et al. Pro-Oncogenic Role of Alternative p38 Mitogen-Activated Protein Kinases p38γ and p38δ, Linking Inflammation and Cancer in Colitis-Associated Colon Cancer. Cancer Res. 2014, 74, 6150–6160.


