1000/1000
Hot
Most Recent
 +1 point
+1 point
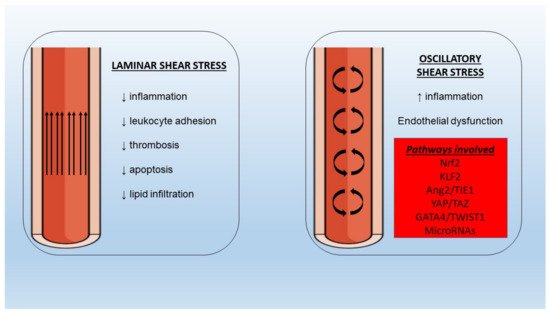
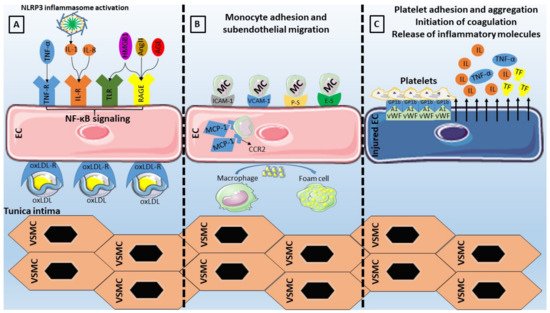
Maintenance of endothelial cell integrity is an important component of human health and disease since the endothelium can perform various functions including regulation of vascular tone, control of hemostasis and thrombosis, cellular adhesion, smooth muscle cell proliferation, and vascular inflammation. Endothelial dysfunction is encompassed by complex pathophysiology that is based on endothelial nitric oxide synthase uncoupling and endothelial activation following stimulation from various inflammatory mediators (molecular patterns, oxidized lipoproteins, cytokines). The downstream signaling via nuclear factor-κB leads to overexpression of adhesion molecules, selectins, and chemokines that facilitate leukocyte adhesion, rolling, and transmigration to the subendothelial space. Moreover, oscillatory shear stress leads to pro-inflammatory endothelial activation with increased monocyte adhesion and endothelial cell apoptosis, an effect that is dependent on multiple pathways and flow-sensitive microRNA regulation. Furthermore, the role of neutrophil extracellular traps and NLRP3 inflammasome as inflammatory mechanisms contributing to endothelial dysfunction has recently been unveiled and is under further investigation.
| Adhesion Molecule | Ligand | Role | Clinical Significance |
|---|---|---|---|
| ICAM-1 | LFA-1 Mac-1 |
Leukocyte adhesion | ICAM-1 correlates with the incidence of CAD and carotid atherosclerosis independently of known cardiovascular risk factors [115]. |
| VCAM-1 | VLA-4 | Leukocyte adhesion | Baseline VCAM-1 is increased in initially healthy middle-aged men who develop cardiovascular disease [116]. |
| E-Selectin | ESL PSGL-1 |
Leukocyte adhesion | E-Selectin correlates with the incidence of CAD and carotid atherosclerosis independently of known cardiovascular risk factors [115]. |
| P-Selectin | PSGL-1 | Leukocyte adhesion | Elevated P-selectin levels predict early adverse events in patients with presumed CAD [117]. |
| MCP-1 | CCR2 | Monocyte chemotaxis | Association of MCP-1 with risk of incident PAD and CAD independently of traditional cardiovascular risk factors [118]. |